
Structures and stabilities of group 17 fluorides EF3 (E = I, At, and element 117) with spin–orbit coupling
Abstract
In this work, a recently developed CCSD(T) approach with spin–orbit coupling (SOC) as well as density functional theory (DFT) using various exchange–correlation (XC) functionals are employed to investigate structures and stabilities of group 17 fluorides EF3 (E = I, At, and element 117). These molecules are predicted to have bent T-shaped C2v structures according to the second-order Jahn–Teller (SOJT) effects or the valance shell electron pair repulsion (VSEPR) theory. For IF3 and (117)F3, our results are consistent with previous SOC-DFT calculations. However, different XC functionals provide different results for AtF3 and our SOC-CCSD(T) calculations show that both the C2v and D3h structures are minima on the potential energy surface and the C2v structure is the global minimum for AtF3. The performance of XC functionals on structures and stabilities of IF3 and AtF3 is found to depend on the fraction of the Hartree–Fock exchange (HFX) included in the XC functionals and the M06-2X functional with 54% of HFX providing results that agree best with CCSD(T) results. In addition, although both the C2v and D3h structures are minima for AtF3, the energy barrier between them is only 8 kJ mol−1 for the C2v structure and 0.05 kJ mol−1 for the D3h structure. This indicates that the D3h structure could not possibly be observed experimentally and AtF3 can convert easily between the three C2v structures. The SOJT term is shown to be reduced by electron correlation for IF3 and AtF3. On the other hand, although SOC decreases the energy difference between the C2v and D3h structures and reduces the deviation of the C2v structure from the D3h structure, it decreases the frequency of the bond bending mode, which may indicate that SOC actually increases the SOJT term. This could be related to
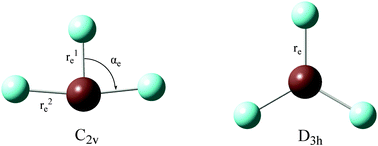
 Please wait while we load your content...
Please wait while we load your content...

