
Double-chain planar D2h B4H2, C2h B8H2, and C2h B12H2: conjugated aromatic borenes†
Abstract
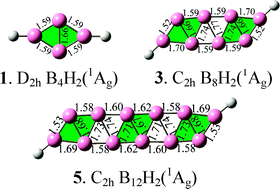
Based upon comprehensive theoretical investigations and known experimental observations, we predict the existence of the double-chain planar D2h B4H2(1), C2h B8H2(3), and C2h B12H2(5) which appear to be the lowest-lying isomers of the systems at the density functional theory level. These conjugated aromatic borenes turn out to be the boron hydride analogues of the conjugated ethylene D2h C2H4(2), 1,3-butadiene C2h C4H6(4), and 1,3,5-hexatriene C2h C6H8(6), respectively, indicating that a B4 rhombus in B2nH2 borenes (n = 2, 4, 6) is equivalent to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond unit in the corresponding CnHn+2 hydrocarbons. Detailed canonical molecular orbital (CMO), adaptive natural density partitioning (AdNDP), and electron localization function (ELF) analyses unravel the bonding patterns of these novel borene clusters and indicate that they are all overall aromatic in nature with the formation of islands of both σ- and π- aromaticity. The double-chain planar or quasi-planar C2v B3H2−(7), C2 B5H2−(8), and C2h B6H2(9) with one delocalized π orbital, C2v B7H2−(10), C2 B9H2−(11), and C2h B10H2(12) with two delocalized π orbitals, and C2v B11H2−(13) with three delocalized π orbitals are found to be analogous in π-bonding to D2h B4H2(1), C2h B8H2(3), and C2h B12H2(5), respectively. We also calculated the electron affinities and ionization potentials of the neutrals and simulated the photoelectron spectroscopic spectra of the monoanions to facilitate their future experimental characterization. The results obtained in this work enrich the analogous relationship between hydroborons and their hydrocarbon counterparts and help to understand the high stability of the theoretically predicted all-boron nanostructures which favor the formation of double-chain substructures.
C double bond unit in the corresponding CnHn+2 hydrocarbons. Detailed canonical molecular orbital (CMO), adaptive natural density partitioning (AdNDP), and electron localization function (ELF) analyses unravel the bonding patterns of these novel borene clusters and indicate that they are all overall aromatic in nature with the formation of islands of both σ- and π- aromaticity. The double-chain planar or quasi-planar C2v B3H2−(7), C2 B5H2−(8), and C2h B6H2(9) with one delocalized π orbital, C2v B7H2−(10), C2 B9H2−(11), and C2h B10H2(12) with two delocalized π orbitals, and C2v B11H2−(13) with three delocalized π orbitals are found to be analogous in π-bonding to D2h B4H2(1), C2h B8H2(3), and C2h B12H2(5), respectively. We also calculated the electron affinities and ionization potentials of the neutrals and simulated the photoelectron spectroscopic spectra of the monoanions to facilitate their future experimental characterization. The results obtained in this work enrich the analogous relationship between hydroborons and their hydrocarbon counterparts and help to understand the high stability of the theoretically predicted all-boron nanostructures which favor the formation of double-chain substructures.
- This article is part of the themed collection: Predicting new molecules by quantum chemical methods
 Please wait while we load your content...
Please wait while we load your content...

