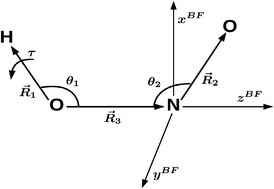
Rovibrational eigenenergies of HONO are computed and compared to experimental energies available in the literature. For their computation, we use a previously developed potential energy surface (PES) and a newly derived exact kinetic energy operator (KEO) including the overall rotation for a tetra-atomic molecule in non-orthogonal coordinates. In addition, we use the Heidelberg Multi-Configuration Time-Dependent Hartree (MCTDH) package. We compare the experimental rovibrational eigenvalues of HONO available in the literature with those obtained with MCTDH and a previously developed potential energy surface (PES) [F. Richter et al., J. Chem. Phys., 2004, 120, 1306.] for the cis geometry. The effect of the overall rotation on the process studied in our previous work on HONO [F. Richter et al., J. Chem. Phys., 2007, 127, 164315.] leading to the cis → transisomerization of HONO is investigated. This effect on this process is found to be weak.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?