
Correction to DFT interaction energies by an empirical dispersion term valid for a range of intermolecular distances†
Abstract
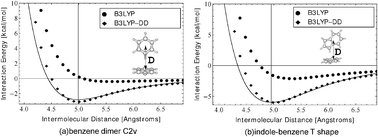
The computation of intermolecular interaction energies via commonly used density functionals is hindered by their inaccurate inclusion of medium and long range dispersion interactions. Practical computation of inter- and intra-macrobiomolecule interaction energies, in particular, requires a fairly accurate yet not overly expensive methodology. It is also desirable to compute intermolecular energies not only at their equilibrium (lowest energy) configurations but also over a range of biophysically relevant distances. We present a method to compute intermolecular interaction energies by including an empirical correction for dispersion which is valid over a range of intermolecular distances. This is achieved by optimizing parameters that moderate the empirical correction by explicit comparison of density functional (B3LYP) energies with distance-dependent (DD) reference values obtained at the CCSD(T)/CBS limit. The resulting method, hereafter referred to as B3LYP-DD, yields interaction energies with an accuracy generally better than 1 kcal mol−1 for different types of noncovalent complexes, over a range of intermolecular distances and interaction strengths, relative to the expensive CCSD(T)/CBS standard. For a training set of dispersion interacting complexes, B3LYP-DD interaction energies in combination with diffuse functions display absolute errors equal to or smaller than 0.68 kcal mol−1. The empirical correction does not significantly increase the computational cost as compared to standard density functional calculations. Applications relevant to biomolecular energy and structure, such as prediction of DNA base-pair interactions, are also presented.
 Please wait while we load your content...
Please wait while we load your content...

