
H-abstraction is more efficient than cis–trans isomerization in (4-methylcyclohexylidene) fluoromethane. An ab initio molecular dynamics study
Abstract
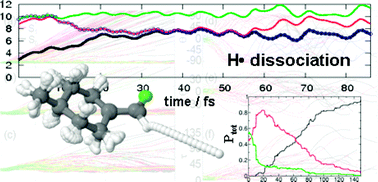
Non-adiabatic molecular dynamics simulations have been performed in the fluoro-olefin (4-methylcyclohexylidene) ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, 4MCF is expected to undergo cis–trans isomerization after light irradiation. However, ab initio molecular dynamics shows that a preferential dissociation of atomic hydrogen is taking place after population transfer to the bright ππ* state. This state is strongly mixed with πσ* states allowing dissociation in the electronic excited state before deactivation to the ground state occurs. A minor amount of trajectories experiences F-dissociation, followed by pyramidalization at the sp2 carbons and CHF dissociation. In contrast, the amount of trajectories undergoing torsion around the double bond, and therefore cis–trans isomerization, is marginal. The H-abstraction reaction is ultrafast, taking place in less than 60 fs.
C double bond, 4MCF is expected to undergo cis–trans isomerization after light irradiation. However, ab initio molecular dynamics shows that a preferential dissociation of atomic hydrogen is taking place after population transfer to the bright ππ* state. This state is strongly mixed with πσ* states allowing dissociation in the electronic excited state before deactivation to the ground state occurs. A minor amount of trajectories experiences F-dissociation, followed by pyramidalization at the sp2 carbons and CHF dissociation. In contrast, the amount of trajectories undergoing torsion around the double bond, and therefore cis–trans isomerization, is marginal. The H-abstraction reaction is ultrafast, taking place in less than 60 fs.
- This article is part of the themed collection: Ultrafast chemical dynamics
 Please wait while we load your content...
Please wait while we load your content...

