
Polarization charge densities provide a predictive quantification of hydrogen bond energies†
Abstract
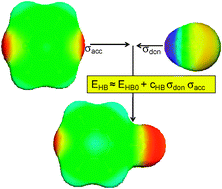
A systematic density functional theory based study of hydrogen bond energies of 2465 single hydrogen bonds has been performed. In order to be closer to liquid phase conditions, different from the usual reference state of individual donor and acceptor molecules in vacuum, the reference state of donors and acceptors embedded in a perfect conductor as simulated by the COSMO solvation model has been used for the calculation of the hydrogen bond energies. The relationship between vacuum and conductor reference hydrogen bond energies is investigated and interpreted in the light of different physical contributions, such as electrostatic energy and dispersion. A very good correlation of the DFT/COSMO hydrogen bond energies with conductor polarization charge densities of separated donor and acceptor atoms was found. This provides a method to predict hydrogen bond strength in solution with a root mean square error of 0.36 kcal mol−1 relative to the quantum chemical dimer calculations. The observed correlation is broadly applicable and allows for a predictive quantification of hydrogen bonding, which can be of great value in many areas of computational, medicinal and physical chemistry.
 Please wait while we load your content...
Please wait while we load your content...

