
Application of a random forests (RF) method as a new approach for variable selection and modelling in a QSRR study to predict the relative retention time of some polybrominated diphenylethers (PBDEs)
Abstract
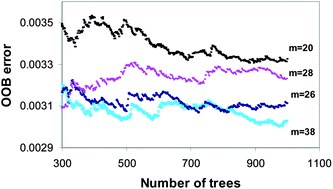
In this work, the method of random forests (RF) was applied for modeling and prediction of the relative retention time of some polybrominated diphenylethers (PBDEs) with descriptors calculated from the molecular structure alone. The effects of tuning parameters such as the number of trees (nt) and the number of randomly selected variables to split each node (m) were investigated. The obtained results showed that the pair (m = 38, nt = 500) can be considered as a plausible setting so that the generalization error was minimal. Also, the importance level of different descriptors was evaluated using RF to simplify the model. The performance of the RF model was compared with the artificial neural network (ANN). Both ANN and RF methods provided accurate predictions, although more accurate results were obtained by the RF model. The determination coefficients of the test set, obtained by the ANN and RF methods, are 0.9619 and 0.9707 respectively.
 Please wait while we load your content...
Please wait while we load your content...

