
Virtual cocrystal screening
Abstract
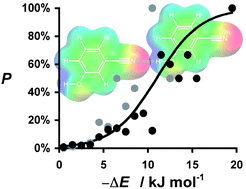
Calculated gas phase molecular electrostatic potential surfaces have been used to identify sets of H-bond donor and H-bond acceptor sites that describe the possible intermolecular interaction sites on the surface of a molecule. The calculated H-bond parameters, αi and βj, were used to estimate interaction site pairing energies in the solid form of the compound through a hierarchical mapping of complementary donor and acceptor sites: the interaction energy for each contact is simply given by the product −αiβj. The approach assumes that all of the interactions that can be made in the solid are made and that the details of three-dimensional structure and crystal packing are of secondary importance. Comparison of the energy of two pure solids with cocrystals of various stoichiometries gives an energy difference, ΔE, which is a measure of the probability of forming a cocrystal. Tests on an experimental cocrystal screen from the literature and the recall of coformers for
 Please wait while we load your content...
Please wait while we load your content...

