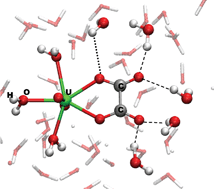
Car–Parrinello molecular dynamics simulations are reported for aqueous UO2(H2O)n(C2O4) (n = 3, 4), calling special attention to the binding modes of oxalate and the thermodynamics of the so-called chelate effect. Based on free energies from thermodynamic integration (BLYP functional), the κ1,κ1′-binding mode of the oxalate (with one O atom from each carboxylate coordinating) is more stable than κ2 (2 O atoms from the same carboxylate) and κ1 forms by 23 and 39 kJ mol−1, respectively. The free energy of binding a fourth water ligand to UO2(H2O)3(κ1-C2O4) is computed to be low, 12 kJ mol−1. Changes of the hydration shell about oxalate during chelate opening are discussed. Composite enthalpies and free energies, obtained from both experiment and quantum-chemical modeling, are proposed for the formation of monodentate UO2(H2O)4(κ1-C2O4). These data suggest that the largest entropy change in the overall complex formation occurs at this stage, and that the subsequent chelate closure under water release is essentially enthalpy-driven.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


