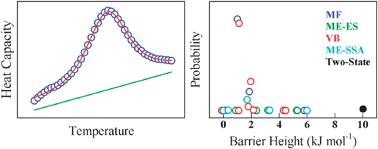
The realization that folding free energy barriers can be small enough to result in significant population of the species at the barrier top has sprouted in several methods to estimate folding barriers from equilibrium experiments. Some of these approaches are based on fitting the experimental thermogram measured by differential scanning calorimetry (DSC) to a one-dimensional representation of the folding free-energy surface (FES). Different physical models have been used to represent the FES: (1) a Landau quartic polynomial as a function of the total enthalpy, which acts as an order parameter; (2) the projection onto a structural order parameter (i.e. number of native residues or native contacts) of the free energy of all the conformations generated by Ising-like statistical mechanical models; and (3) mean-field models that define conformational entropy and stabilization energy as functions of a continuous local order parameter. The fundamental question that emerges is how can we obtain robust, model-independent estimates of the thermodynamic folding barrier from the analysis of DSC experiments. Here we address this issue by comparing the performance of various FES models in interpreting the thermogram of a protein with a marginal folding barrier. We chose the small α-helical protein PDD, which folds–unfolds in microseconds crossing a free energy barrier previously estimated as ∼1 RT. The fits of the PDD thermogram to the various models and assumptions produce FES with a consistently small free energy barrier separating the folded and unfolded ensembles. However, the fits vary in quality as well as in the estimated barrier. Applying Bayesian probabilistic analysis we rank the fit performance using a statistically rigorous criterion that leads to a global estimate of the folding barrier and its precision, which for PDD is 1.3 ± 0.4 kJ mol−1. This result confirms that PDD folds over a minor barrier consistent with the downhill folding regime. We have further validated the multi-model Bayesian approach through the analysis of two additional protein systems: gpW, a midsize single-domain with α + β topology that also folds in microseconds and has been previously catalogued as a downhill folder, and α-spectrin SH3, a domain of similar size but with a β-barrel fold, slow-folding kinetics and two-state-like thermodynamics. From a general viewpoint, the Bayesian analysis developed here results in a statistically robust, virtually model-independent, method to estimate the thermodynamic free-energy barriers to protein folding from DSC thermograms. Our method appears to be sufficiently accurate to consistently detect small differences in the barrier height, and thus opens up the possibility of characterizing experimentally the changes in thermodynamic folding barriers induced by single-point mutations on proteins within the downhill regime.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


