
New electron correlation theories for transition metal chemistry
Abstract
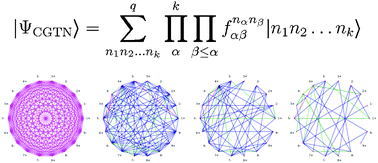
Electronic structure theory faces many computational challenges in transition metal chemistry. Usually, density functional theory is the method of choice for theoretical studies on transition metal complexes and clusters mostly because it is the only feasible one, although its results are not systematically improvable. By contrast, multireference ab initio methods could provide a correct description of the electronic structure, but are limited to small molecules because of the tremendous computational resources required. In recent years, conceptually new ab initio methods emerged that turned out to be promising for theoretical coordination chemistry. We review and discuss two efficient parametrization schemes for the electronic wave function, the matrix product states and the complete-graph tensor network states. Their advantages are demonstrated at example transition metal complexes. Especially, tensor network states might provide the key to accurately describe strongly correlated and magnetic molecular systems in transition metal chemistry.
 Please wait while we load your content...
Please wait while we load your content...

