
Molecular dynamics effects on luminescence properties of oligothiophene derivatives: a molecular mechanics–response theory study based on the CHARMM force field and density functional theory
Abstract
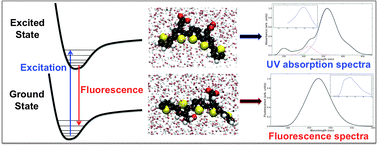
CHARMM force field parameter values for a class of oligothiophene derivatives have been derived with reference to density functional theory/B3LYP potential energy surfaces. The force field parametrization of these luminescent conjugated polyelectrolytes includes the electronic ground state as well as the strongly light absorbing first excited state. In conjunction with quantum chemical response theory calculations of transition state properties, a molecular dynamical model of the Stokes shift is obtained. The theoretical model is benchmarked against
 Please wait while we load your content...
Please wait while we load your content...

