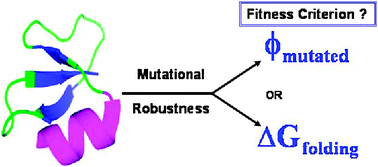
This work explores the effects of two different fitness criteria, the free energy of folding (ΔGfolding) and foldability (ϕ) of mutated sequences, by measuring the designed protein's robustness via cumulative random point mutations. The results of a self-consistent mean-field based theory are used to design ‘wild type’ protein sequences corresponding to a specified target structure for a given foldability criteria ϕ. The theory is applied on three 36-mer real protein conformations and the ‘wild type’ sequences are identified in terms of site specific monomer probabilities corresponding to a given foldability. Unlike the stability criteria, ΔGfolding < 0, the foldability criteria ϕmutated < −1 effectively identifies sequences of different site-specific monomer identities by specifying the mean and variance of the energy of the unfolded state ensemble. The results depict a distinct difference in the pattern of mutational robustness of the neutral sequence space, ϕmutated < −1 scans more number of neutral sequences compared to ΔGfolding < 0 to find the evolutionary fit sequences. ϕmutated < −1 also accounts for marginally stable sequences which are not effectively scanned by ΔGfolding < 0 to determine evolutionary fitness. The results clearly point out that viable mutated sequences that are foldable, may not always conform to ΔGfold < 0, hence assessing the role of foldability in addition to stability for determining protein's robustness towards cumulative random point mutations. These observations may be used in engineering and designing de novoprotein sequences which are more robust towards random point mutations.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


