
Modelling molecule–surface interactions—an automated quantum-classical approach using a genetic algorithm
Abstract
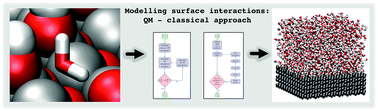
We present an automated and efficient method to develop force fields for molecule–surface interactions. A genetic algorithm (GA) is used to parameterise a classical force field so that the classical adsorption energy landscape of a molecule on a surface matches the corresponding landscape from density functional theory (DFT) calculations. The procedure performs a sophisticated search in the parameter phase space and converges very quickly. The method is capable of fitting a significant number of structures and corresponding
- This article is part of the themed collection: Multiscale modelling
 Please wait while we load your content...
Please wait while we load your content...

