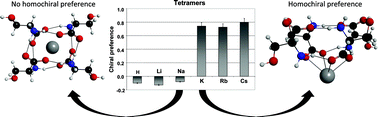
Serine “magic-number” clusters have attracted substantial experimental and theoretical interest since their discovery. Serine undergoes marked chiral enrichment upon sublimation, which has been associated with the homochiral selectivity of the octamer. This process has been implicated in one possible mechanism leading to the origin of biological homochirality. While the octamer is the best known of the serine clusters, here we focus on the tetramer, the smallest serine cluster known to exhibit homochiral preference. This choice is based on its greater simplicity and tractability with accessible computational resources. Basin-hopping molecular dynamics simulations coupled to density functional theory calculations yield a “structural landscape” for low-lying configurations on the potential energy surface. The full range of enantiomeric compositions and charge states is investigated. Global energy minimum serine tetramers consist of a cage structure bonded by zwitterionic terminal groups. The participation of the serine hydroxyl side chains in hydrogen bonds with adjacent monomers drives the homochiral selectivity of serine tetramers. The configuration of the hydrogen bonding network is strongly dependent on enantiomeric composition and charge state. Smaller cations are incorporated into the center of the tetramer cage and effectively disable all side chain hydrogen bonding, while larger cations appear not to incorporate into the tetramer cage and are stabilized outside only in the homochiral case. The current theoretical data requires the introduction of a kinetic barrier to complete the model, limiting rearrangement from the basic cage configuration in some cases, which is discussed and probed directly by doubly-nudged elastic band transition state searches. These calculations elucidate a large barrier for reorganization of the cage, completing the theoretical understanding of the tetramers.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


