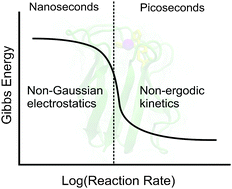
Despite its diversity, life universally relies on a simple basic mechanism of energy transfer in its energy chains—hopping electron transport between centers of electron localization on hydrated proteins and redox cofactors. Since many such hops connect the point of energy input with a catalytic site where energy is stored in chemical bonds, the question of energy losses in (nearly activationless) electron hops, i.e., energetic efficiency, becomes central for the understanding of the energetics of life. We show here that standard considerations based on rules of Gibbs thermodynamics are not sufficient, and the dynamics of the protein and the protein–water interface need to be involved. The rate of electronic transitions is primarily sensitive to the electrostatic potential at the center of electron localization. Numerical simulations show that the statistics of the electrostatic potential produced by hydration water are strongly non-Gaussian, with the breadth of the electrostatic noise far exceeding the expectations of the linear response. This phenomenon, which dramatically alters the energetic balance of a charge-transfer chain, is attributed to the formation of ferroelectric domains in the protein's hydration shell. These dynamically emerging and dissipating domains make the shell enveloping the protein highly polar, as gauged by the variance of the shell dipole which correlates with the variance of the protein dipole. The Stokes-shift dynamics of redox-active proteins are dominated by a slow component with the relaxation time of 100–500 ps. This slow relaxation mode is frozen on the time-scale of fast reactions, such as bacterial charge separation, resulting in a dramatically reduced reorganization free energy of fast electronic transitions. The electron transfer activation barrier becomes a function of the corresponding rate, self-consistently calculated from a non-ergodic version of the transition-state theory. The peculiar structure of the protein–water interface thus provides natural systems with two “non's”—non-Gaussian statistics and non-ergodic kinetics—to tune the efficiency of the redox energy transfer. Both act to reduce the amount of free energy released as heat in electronic transitions. These mechanisms are shown to increase the energetic efficiency of protein electron transfer by up to an order of magnitude compared to the “standard picture” based on canonical free energies and the linear response approximation. In other words, the protein–water tandem allows both the formation of a ferroelectric mesophase in the hydration shell and an efficient control of the energetics by manipulating the relaxation times.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


