
A density functional theory approach to noncovalent interactions via interacting monomer densities
Abstract
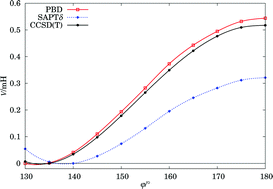
A recently proposed “DFT + dispersion” treatment (Rajchel et al., Phys. Rev. Lett., 2010, 104, 163001) is described in detail and illustrated by more examples. The formalism derives the dispersion-free density functional theory (DFT) interaction energy and combines it with the dispersion energy from separate DFT calculations. It consists of the self-consistent polarization of DFT monomers restrained by the exclusion principle via the Pauli blockade technique. Within the monomers a complete exchange-correlation potential should be used, but between them only the exact exchange operates. The application to a wide range of molecular complexes from rare-gas dimers to hydrogen-bonds to π-electron interactions shows good agreement with benchmark values.
 Please wait while we load your content...
Please wait while we load your content...

