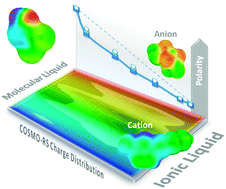
A COSMO-RS descriptor (Sσ-profile) has been used in quantitative structure–property relationship (QSPR) studies by a neural network (NN) for the prediction of empirical solvent polarity ENT scale of neat ionic liquids (ILs) and their mixtures with organic solvents. Sσ-profile is a two-dimensional quantum chemical parameter which quantifies the polar electronic charge of chemical structures on the polarity (σ) scale. Firstly, a radial basis neural network exact fit (RBNN) is successfully optimized for the prediction of ENT, the solvatochromic parameter of a wide variety of neat organic solvents and ILs, including imidazolium, pyridinium, ammonium, phosphonium and pyrrolidinium families, solely using the Sσ-profile of individual molecules and ions. Subsequently, a quantitative structure–activity map (QSAM), a new concept recently developed, is proposed as a valuable tool for the molecular understanding of IL polarity, by relating the ENT polarity parameter to the electronic structure of cations and anions given by quantum-chemical COSMO-RS calculations. Finally, based on the additive character of the Sσ-profile descriptor, we propose to simulate the mixture of IL–organic solvents by the estimation of the SMixtureσ-profile descriptor, defined as the weighted mean of the Sσ-profile values of the components. Then, the ENT parameters for binary solvent mixtures, including ILs, are accurately predicted using the SMixtureσ-profile values from the RBNN model previously developed for pure solvents. As result, we obtain a unique neural network tool to simulate, with similar reliability, the ENT polarity of a wide variety of pure ILs as well as their mixtures with organic solvents, which exhibit significant positive and negative deviations from ideality.

 Please wait while we load your content...
Please wait while we load your content...

