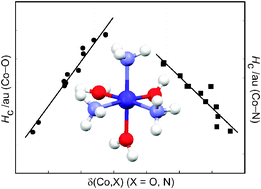
Spin-unrestricted DFT-X3LYP/6-311++G(d,p) calculations have been performed on a series of complexes of the form [Co(H2O)6−n(NH3)n]2+ (n = 0–6) to examine their equilibrium gas-phase structures, energetics, and electronic properties in their quartet electronic ground states. In all cases Co2+ in the energy-minimised structures is in a pseudo-octahedral environment. The calculations overestimate the Co–O and Co–N bond lengths by 0.04 and 0.08 Å, respectively, compared to the crystallographically observed mean values. There is a very small Jahn–Teller distortion in the structure of [Co(H2O)6]2+ which is in contrast to the very marked distortions observed in most (but not all) structures of this cation that have been observed experimentally. The successive replacement of ligated H2O by NH3 leads to an increase in complex stability by 6 ± 1 kcal mol−1 per additional NH3 ligand. Calculations using UB3LYP give stabilisation energies of the complexes about 5 kcal mol−1 smaller and metal–ligand bond lengths about 0.005 Å longer than the X3LYP values since the X3LYP level accounts for the London dispersion energy contribution to the overall stabilisation energy whilst it is largely missing at the B3LYP level. From a natural population analysis (NPA) it is shown that the formation of these complexes is accompanied by ligand-to-metal charge transfer the extent of which increases with the number of NH3 ligands in the coordination sphere of Co2+. From an examination of the topological properties of the electron charge density using Bader’s quantum theory of atoms in molecules it is shown that the electron density ρc at the Co–O bond critical points is generally smaller than that at the Co–N bond critical points. Hence Co–O bonds are weaker than Co–N bonds in these complexes and the stability increases as NH3 replaces H2O in the metal’s coordination sphere. Several indicators, including the sign and magnitude of the Laplacian of the charge density ∇2ρc, the ratio of the local potential and kinetic energy densities, |Vc|/Gc, the sign of the total energy density Hc, and the delocalisation index δ(Co,X), X = O, N, are used to show that whilst the metal–ligand bonds are predominantly ionic in nature, they gain covalent character as NH3 replaces H2O, and the Co–N bond is significantly more covalent than the Co–O bond. We have shown that the delocalisation index δ(Co,X), X = O, N, is strongly correlated with the zero-point corrected stabilisation energy Ecd demonstrating that δ can be used as a measure of the bond stability in these complexes.

 Please wait while we load your content...
Please wait while we load your content...

