Theoretical calculations and in situ solid state NMR spectroscopy have been combined to get insight on the nature of the active sites for the Beckmann rearrangement reaction in borosilicate zeolites. The interaction of a B site in zeolite Beta with a series of probe molecules (ammonia, pyridine, acetone and water) has been modelled and the 15N and 11B NMR isotropic chemical shift of the resulting complexes calculated and compared with experimental in situ NMR results. This approach has allowed validation of the methodology to model the adsorption on a zeolite boron site of molecules of varying basicity which are either protonated or non-protonated. The limitation is that theoretical calculations overestimate the effect of molecular adsorption through hydrogen bonds on the calculated isotropic 11B NMR chemical shift.
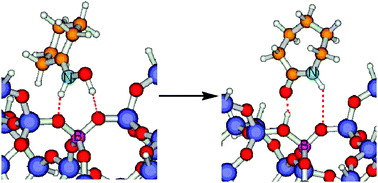
Theoretical and experimental results on the adsorption of acetophenone and cyclohexanone oximes on zeolite B-Beta indicate that Brønsted acid sites protonate the oximes, changing the boron coordination from trigonal to tetrahedral. Comparison of theoretical and experimental 15N NMR chemical shifts of the adsorbed amides (acetanilide and ε-caprolactam) indicates that they are non-protonated, and the 11B NMR spectra show that, as expected, boron remains in trigonal coordination with an isotropic δ 11Bexp which differs from the calculated value δ 11Bcalc.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


