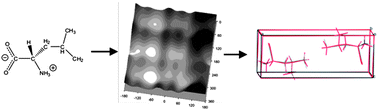
Crystal structure prediction calculations are performed for four hydrophobic amino acids (alanine, valine, leucine and isoleucine), to test the computational methods that have been developed for flexible organic molecules. Specific focus is placed on the final energy minimisation and optimisation of the molecular conformations in the computer-generated crystal structures. Overall, the results are very encouraging. The observed crystal structures are usually found as the lowest energy predicted structures, demonstrating that crystal packing is predictable by computational methods, even for fairly challenging systems. In addition to the assessment of the computational methods, comparison of the hypothetical with the observed crystal structures provides insight into the balance between hydrogen bonding and hydrophobic side-chain packing that determines the crystal structures of these biologically important molecules.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?