
Computational study through atomistic potentials of a partial bilayer liquid crystal: structure and dynamics
Abstract
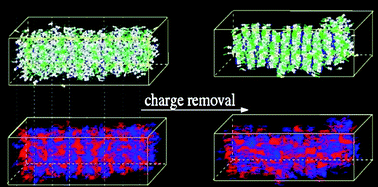
An ab initio derived force-field, previously (Cacelli et al., J. Phys. Chem. B, 2007, 111, 2130) tested and validated on the structural and dynamic properties of isotropic and nematic phases of the 4-alkyl-4′-cyanobiphenyl (nCB; n = 5–8) series, is employed here in molecular dynamics simulations to verify the model’s capability of reproducing the experimental partial double layer smectic phase (SmAd) shown by the 4-octyl-4′-cyanobiphenyl homologue. Lengthy (≃50 ns) NPT simulations, at 1 atm and 300 K, have been performed and the resulting mesophase was found to be in good agreement with most of the considered experimental data. Furthermore, by comparing the results obtained with systems of different size, the effect of both the number of molecules and smectic layers on mesophase stability is discussed. Particular attention has been also paid to the role of dipolar interactions within the double layer, by comparing the results obtained employing the force field with and without point charges. In the latter case, the SmAd phase was found to collapse into a single layer smectic phase. Finally dynamic properties have been computed from NVE trajectories and compared with their experimental counterparts.
 Please wait while we load your content...
Please wait while we load your content...

