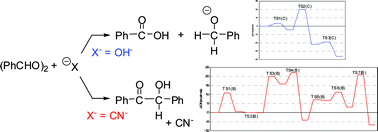
The Cannizzaro reaction (“RX C”) and the benzoin reaction (“RX B”) were investigated by density functional theory calculations. Reaction models (benzaldehyde)2 + X− + (H2O)n [for X− = OH− (RX C) n = 8, and for X− = CN− (RX B) n = 8 or n = 14] were adopted. Three transition states (TSs) were obtained for RX C, and the rate-determining step was confirmed to be the hydride shift. The single electron transfer path was also obtained, which is supported by the C–O−⋯CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O attraction. In RX B, seven TSs were obtained. The C⋯C bond formation TS, TS4(B), was found to be the rate-determining step. However, the carbanion-formation TS (TS3(B)) and the CN− release TS (TS7(B)) are also of large activation free energies (ΔG‡s). The result ΔG‡(TS4(B)) ≥ΔG‡(TS7(B)) ∼ΔG‡(TS3(B)) was obtained with both n = 8 and n = 14 models. Proton relays along the linear hydrogen bonds are concerned with bond interchanges and promote well arranged and successive elementary processes in RXs C and B.
O attraction. In RX B, seven TSs were obtained. The C⋯C bond formation TS, TS4(B), was found to be the rate-determining step. However, the carbanion-formation TS (TS3(B)) and the CN− release TS (TS7(B)) are also of large activation free energies (ΔG‡s). The result ΔG‡(TS4(B)) ≥ΔG‡(TS7(B)) ∼ΔG‡(TS3(B)) was obtained with both n = 8 and n = 14 models. Proton relays along the linear hydrogen bonds are concerned with bond interchanges and promote well arranged and successive elementary processes in RXs C and B.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?