
Site preference of Eu2+ dopants in the (Ba,Sr)13−xAl22−2xSi10+2xO66 phosphor and its effect on the luminescence properties: a density functional investigation
Abstract
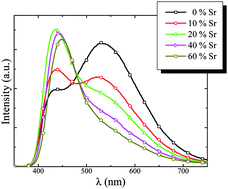
First principles density functional calculations were carried out for model compounds Ba12EuAl22Si10O66 and Sr12EuAl22Si10O66 to account for the luminescence properties of Eu2+ doped Ba13−xAl22−2xSi10+2xO66 and Eu2+ doped (Ba,Sr)13−xAl22−2xSi10+2xO66. Both host lattices have three different crystallographic alkaline earth sites, namely, AE(1), AE(2) and AE(3), at which the dopants Eu2+ are located. The fluorescence properties of Eu2+ doped Ba13−xAl22−2xSi10+2xO66 are not explained if the dopant ions Eu2+ are considered to randomly occupy the three different Ba sites, but are explained if the dopants are considered to preferentially occupy the Ba(3) sites. In contrast, the fluorescence properties of Eu2+ doped (Ba,Sr)13−xAl22−2xSi10+2xO66 are explained only when both Sr2+ and Eu2+
 Please wait while we load your content...
Please wait while we load your content...