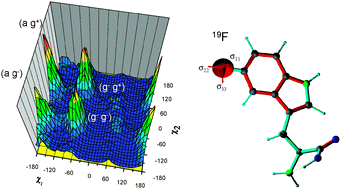
A semi-empirical method for the prediction of chemical shifts, based on bond polarization theory, has recently been introduced for 13C. Here, we extended this approach to calculate the 19F chemical shift tensors of fluorine bound to aromatic rings and in aliphatic CF3groups. For the necessary parametrization, ab initio chemical shift calculations were performed at the MP2 level for a set of fluorinated molecules including tryptophan. The bond polarization parameters obtained were used to calculate the 19F chemical shift tensors for several crystalline molecules, and to reference the calculated values on a chemical shift scale relative to CFCl3. As a first biophysical application, we examined the distribution of conformations of a 19F-labeled tryptophan side chain in the membrane-bound ion channel peptide, gramicidin A. The fluorine chemical shift tensors were calculated from snapshots of a molecular dynamics simulation employing the 19F-parametrized bond polarization theory. In this MD simulation, published 2H quadrupolar and 15N–1H dipolar couplings of the indole ring were used as orientational constraints to determine the conformational distribution of the 5F-Trp13 side chain. These conformations were then used to interpret the spectra of 19F-labeled gramicidin A in fluid and gel phase lipid bilayers.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


