
Molecular dynamics study of the silica–water–SDA interactions
Abstract
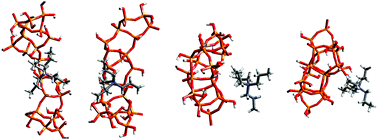
In this paper we have applied the molecular dynamics simulations in order to analyse the role of the structure directing tetrapropylammonium ions in the aggregation process that leads to silicalite formation. We address the specific question of how the interactions between
 Please wait while we load your content...
Please wait while we load your content...

