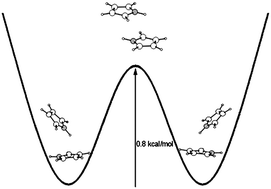
The energy, dynamical geometry characteristics and low frequency intermolecular vibrations of the pyrrole dimer have been examined at the MP2 and CCSD(T) levels of ab initio theory. The actual distortions of the pyrrole dimer from its reference (equilibrium) position were measured using the distance of the monomer mass centres (R), the angle between their planes (the mirror planes orthogonal to the molecular planes of both monomers were assumed to coincide) and the angle between the R directional vector and the proton-accepting monomer plane; the structures of the monomers were assumed to be unchanged by dimerisation. The lowest part of the potential energy function confining the probed motions possessed two equivalent energy pockets with the CCSD(T)/complete basis set limit stabilisation energy of 6.2 kcal mol−1 separated by a relatively low barrier (0.8 kcal mol−1), thus raising questions concerning the classical interconversion of the T-shaped equilibrium structures via a C2h parallel-displaced transient structure and/or quantum mechanical tunnellings through the barrier. The questions have been answered unequivocally by calculating the energies and tunnelling splittings of the relevant vibrational levels. Importantly: (a) all the excited tunnelling (interconverting) states underwent fast geometry interconversions, hence evidencing conformational instability of the studied dimer under usual laboratory conditions; (b) the dynamical averages of the used geometry characteristics exhibited profound tunnelling (interconverting) dependences, thus advocating that they be respected in reliable structural studies of the pyrrole dimer and chemically similar systems; (c) the geometry characteristics of the ground vibrational state agreed quite reasonably with their experimental counterparts, evidencing the adequacy of the theory used and the reliability of the characteristics predicted for the excited vibrational states; and (d) the calculated dissociation barrier of the dimer exceeds its experimentally derived analogue by more than three times, showing the inadequacy of the constraining assumptions used to derive it from the experimental spectra.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


