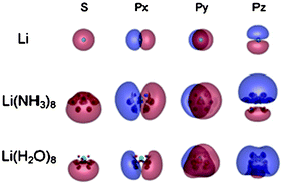
A theoretical study of the ground and low-lying excited states of Li(NH3)n and Li(H2O)n (n = 1–8) clusters is presented. Their structures, binding energies, vertical ionization energies and vertical transition energies were calculated using ab initio molecular orbital methods at correlated levels. Compared with Na(NH3)n and Na(H2O)n, the incremental binding energies and the spectroscopic energies are found to be almost metal-independent, but solvent-dependent after first-shell closure in both M(NH3)n and M(H2O)n (M = Li and Na) clusters. Autoionization of the alkali atoms occurs via a spatial expansion of unpaired electron distribution extending to outside of the first solvation-shell, irrespective of the combinations of the metal and the solvent. The localization mode of the wave function of the solvated electron was investigated in both the ground and excited states. A change from a one-center diffuse state to a two-center localized state proceeds more quickly against n in M(H2O)n than in M(NH3)n, which is behind the solvent-dependence of the evaluated quantities.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


