
Formation entropies of intrinsic point defects in cubic In2O3 from first-principles density functional theory calculations
Abstract
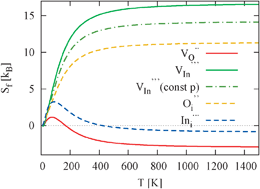
Entropy contributions to the Gibbs free energy of defect formation of vacancies and interstitials in cubic In2O3 are calculated by means of first-principles calculations. We employ the supercell formalism together with a pseudo-potential and plane-wave based density functional method for the force calculations. Our results suggest that temperature-dependent contributions to the Gibbs free energies of defect formation can rise to 1.4 eV at 1000 K and therefore cause variations in the predicted defect equilibria as compared to calculations based on static energy data. We thoroughly discuss elastic contributions to the defect formation entropy at constant volume or pressure and address the correct treatment of an entropy reservoir for a binary system. Finally, we investigate the temperature dependence of the defect formation entropy and compare this to the usual high-temperature approximation.
- This article is part of the themed collection: Physical chemistry of solids - The science behind materials engineering
 Please wait while we load your content...
Please wait while we load your content...

