
Classical, quantum and statistical simulations of vibrationally excited HOSO2: IVR, dissociation, and implications for OH + SO2 kinetics at high pressures
Abstract
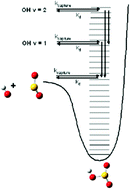
In this paper, we present classical and coupled coherent states quantum dynamics simulations to investigate intramolecular vibrational energy redistribution (IVR) from an excited (v = 1–10) OH stretch within the HOSO2 complex to the other molecular bath modes. Using an analytical PES derived from electronic structure theory calculations, the results obtained from both the classical and quantum simulations are in reasonable agreement. The dynamics results suggest that statistical models overpredict HOSO2 dissociation k(E)s, and underpredict the amount of vibrational excitation in the nascent OH formed following complex dissociation. In order to understand the dynamics results, we utilize a simple analytical model for describing energy flow from excited modes to bath modes, and show that IVR limits complex dissociation at short times. We also consider qualitative mass affects on IVR, and consider the implications of this work on previous measurements of the OH + SO2 association k∞ using the proxy method.
 Please wait while we load your content...
Please wait while we load your content...

