
A ground-state-directed optimization scheme for the Kohn–Sham energy†
Abstract
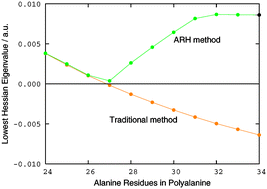
Kohn–Sham density-functional calculations are used in many branches of science to obtain information about the electronic structure of molecular systems and materials. Unfortunately, the traditional method for optimizing the Kohn–Sham energy suffers from fundamental problems that may lead to divergence or, even worse, convergence to an energy saddle point rather than to the ground-state minimum—in particular, for the larger and more complicated electronic systems that are often studied by Kohn–Sham theory nowadays. We here present a novel method for Kohn–Sham energy minimization that does not suffer from the flaws of the conventional approach, combining reliability and efficiency with linear complexity. In particular, the proposed method converges by design to a minimum, avoiding the sometimes spurious solutions of the traditional method and bypassing the need to examine the structure of the provided solution.
 Please wait while we load your content...
Please wait while we load your content...

