
Real wave packet and quasiclassical trajectory studies of the H+ + LiH reaction
Abstract
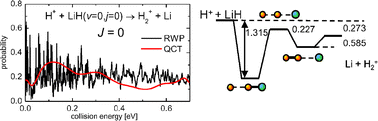
Time-dependent real wave packet (RWP) and quasiclassical trajectory (QCT) calculations have been carried out to study the H+ + LiH reaction on the ab initio potential-energy surface of Martinazzo et al. [J. Chem. Phys., 2003, 119, 11241]. Total initial state-selected and final state-resolved reaction probabilities for the two possible reaction channels, H2+ + Li and LiH + H+, have been calculated for total angular momentum J = 0 at a broad range of collision energies. Integral cross sections and thermal rate coefficients have been calculated using the QCT method and from the corresponding J = 0 RWP reaction probabilities by means of a capture model. The calculated thermal rate coefficients are found to be nearly independent of temperature in the 100–500 K interval with a value of ≈10−9 cm3 s−1, which is in good agreement with estimates used in evolutionary models of early-Universe lithium chemistry. The RWP results are found to be in good agreement overall with the corresponding QCT calculations.
 Please wait while we load your content...
Please wait while we load your content...

