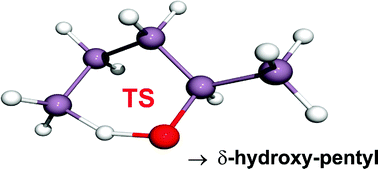
Theoretical models have been used to derive rate coefficients for the unimolecular reaction pathways of two prototypical alkoxyl radicals (1-butoxyl and 2-pentoxyl) which can undergo 1,5 H-shift isomerisation yielding the corresponding δ-hydroxy alkyl radical. Special emphasis has been given to the contribution of tunnelling in the isomerisation channels which has not been accounted for in previous theoretical studies. The combination of high level ab initio calculations with a fully coupled multiple channel master equation (ME) treatment resulted in a significant increase of the isomerisation rates by about a factor of 2.7 for the 1-butoxyl and 2.3 for the 2-pentoxyl radical, respectively, as compared to calculations in which tunnelling was neglected, even at 298 K. The corresponding Arrhenius energies in the temperature range from 200 up to 1000 K are significantly smaller when tunnelling is accounted for and differ from previous results which focused only on temperatures around 298 K. The rate expressions derived for the 1,5 H-shift isomerisation reactions are: kiso,1but = 1.58 × 1012(T/300 K)−2.30 exp(−4679 K/T) s−1 and kiso,2pent = 4.65 × 1012(T/300 K)−3.22 exp(−4782 K/T) s−1 valid for p = 1013 mbar and temperatures between 200 K ≤ T ≤ 1000 K. Our results are strongly supported by recent experiments of Cox and co-workers, (D. Johnson, P. Cassanelli, and R. A. Cox, J. Phys. Chem. A, , 2004, 108, 519 and P. Cassanelli, D. Johnson, and R. A. Cox, Phys. Chem. Chem. Phys. 2005, 7, 3702, 18) which are among the very few studies performed in a temperature regime (250 ≤ T ≤ 320 K) where tunnelling is thought to be effective. Since tunnelling has been neglected so far in the theoretical analysis of experimental data, the reliability of existing extrapolations to higher and lower temperatures is discussed in detail.

 Please wait while we load your content...
Please wait while we load your content...

