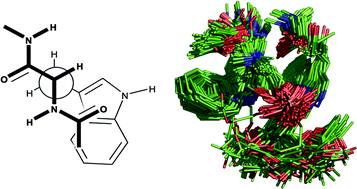
A series of molecular models of the adducts formed between N-acetyl-L-tryptophan ethylamide and diacetyl-sn-glycero-3-phosphocholine have been generated. Using rOesy data that enabled us to place restrictions on the proximity of a number of key protons in the amino acid/phosphocholine pairs, a series of structures were generated following molecular dynamics and mechanics experiments using the CHARMM27 force field. These structures were then subjected to a series of clustering algorithms in order to classify the tight binding interactions between a single tryptophan and a phosphocholine. From these analyses, it is evident that: (i) binding is characterised by hydrogen bonding between the indole NH as donor and phosphate oxygen as acceptor, cation–carbonyl interactions between the cholineammonium and amide carbonyl groups and cation–π interactions; (ii) cation–π interactions are not always observed, particularly when their formation is at the expense of cation–carbonyl and hydrogen bonding interactions; (iii) on the basis of amino acid torsional parameters, it is possible to predict whether the phosphocholine headgroup will bind in a compact or elongated conformation. Extension of the procedures to characterise 2 : 1 Trp–PC binding revealed that the same intermolecular interactions are predominant; however, combinations of all three intermolecular interactions within the same adduct occur much more frequently due to the availability of donor/acceptor groups from both tryptophans in the 2 : 1 system.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


