
A coupled cluster study of the electronic spectroscopy and photochemistry of Cr(CO)6†
Abstract
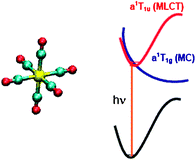
The transition energies to the low-lying singlet and triplet excited states of Cr(CO)6 are computed by equation-of-motion coupled cluster singles and doubles (EOM-CCSD) and similarity transformed equation-of-motion coupled cluster singles and doubles (STEOM-CCSD) methods with all-electrons basis sets. Both experimental and optimized geometries are used for the calculations. Calculations with various basis sets, among them one of the largest calculations performed at the EOM-CCSD level, based on atomic natural orbitals with 627 functions, were used to evaluate the basis set influence on computed transition energies. The presence of a shoulder at 3.9 eV in the experimental absorption spectrum, assigned to the 1A1g
→
1T2u transition, which was not reproduced by recent density functional theory (DFT) or multi-state complete active space perturbation theory (MS-CASPT2) is supported by the present STEOM-CCSD calculations with a theoretical value of 3.92 eV. In addition to this weak 1A1g
→ a 1T2u absorption, we observe two strong absorptions corresponding to 1A1g
→ a 1T1u at 4.37 eV (vs. an experimental value of 4.46 eV) and 1A1g
→ b 1T1u at 5.20 eV (vs. an experimental value of 5.53 eV). Both are characterized as metal-to-
 Please wait while we load your content...
Please wait while we load your content...

