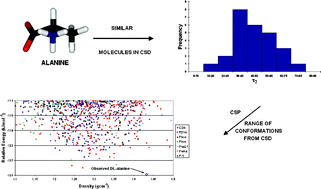
Crystal structure prediction calculations have been performed for the α-amino acid alanine with the intention of developing reliable computational methods for flexible molecules and, specifically, to study the crystal packing of the more flexible amino acids. For the α-amino acids, the density functional theory geometry optimised conformations of the isolated molecules are considerably different, in both geometry and form, to what is observed in the crystal structures. The molecules take the zwitterionic form in the observed crystals, but are nonionised for the isolated molecules. The quantum mechanically optimised structure of the isolated molecule is therefore a poor starting point for computationally generating putative crystal structures. We show that, by limiting the conformations of alanine to the torsion angle distributions in the observed crystal structures of similar molecules in the Cambridge Structural Database, sets of likely crystal structures can be generated, with the lowest energy racemic and enantiopure crystal structures corresponding to the experimentally observed crystal structures.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


