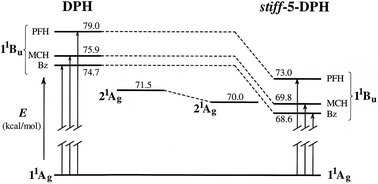
all-trans-1,4-Diindanylidenyl-2-butene (ttt-stiff-5-DPH), a torsionally constrained analogue of all-trans-1,6-diphenyl-1,3,5-hexatriene (ttt-DPH), was synthesized and studied in order to evaluate the role of phenyl–vinyl torsional motions in the photophysical and photochemical responses of the DPH chromophore. Spectroscopic and photoisomerization measurements reveal that the behavior of the rigid DPH analogue is very similar to that of the parent DPH. This similarity is obtained despite the fact that the alkyl substitution from the five-membered rings selectively lowers the energy of the 1 1Bu* state, leading to inversion of the order of the 1 1Bu* and 2 1Ag* energy levels in hydrocarbon solvents. In stiff-5-DPH, as in DPH, an increase in solvent polarity enhances terminal over central bond photoisomerization. Analyses of fluorescence and photoisomerization quantum yields show that, as in DPH, the torsional relaxation channel on the singlet excited state manifold is inefficient, falling far short of accounting for all radiationless decay. Significant (∼50 and 80% of all singlet decay in Bz and AN, respectively), photochemically unproductive, radiationless decay channels exist in both molecules. Competing one bond photoisomerizations give the two major photoproducts: tct-stiff-5-DPH and ctt-stiff-5-DPH. They were isolated in pure form and were spectroscopically characterized. Biacetyl-sensitization was used to study the behavior of the stiff-5-DPH triplet state. As in the parent DPH, stiff-5-DPH triplets undergo relatively efficient concentration dependent geometric photoisomerization.

 Please wait while we load your content...
Please wait while we load your content...