
An ab initio investigation of the O(3P)–H2(1Σ +g) van der Waals well
Abstract
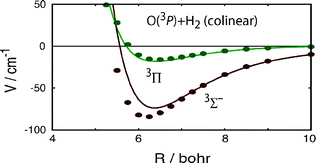
We report an ab initio study of the van der Waals region of the O(3P)–H2 potential energy surface based on RCCSD(T) calculations with an aug-cc-pVQZ basis supplemented by bond functions. In addition, an open-shell implementation of symmetry-adapted perturbation theory (SAPT) is used to corroborate the RCCSD(T) calculations and to investigate the relative magnitudes of the various contributions to the van der Waals interaction. We also investigate the effect of the spin–orbit coupling on the position and depth of the van der Waals well. We predict the van der Waals minimum to occur in perpendicular geometry, and located at a closer distance than a secondary well in colinear geometry. The potentials obtained in the present study confirm the previous calculations of Alexander [M. H. Alexander, J. Chem. Phys., 1998, 108, 4467], but disagree with the earlier work of Harding and co-workers [Z. Li, V. A. Apkarian and L. B. Harding, J. Chem. Phys., 1997, 106, 942] as well as with recently refitted surfaces of Brandão and coworkers [J. Brandão, C. Mogo and B. C. Silva, J. Chem. Phys., 2004, 121, 8861]. Inclusion of spin–orbit coupling reduces the depth of the van der Waals minimum without causing a change in its position.
 Please wait while we load your content...
Please wait while we load your content...

