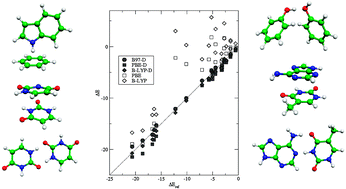
Density functional theory including dispersion corrections (DFT-D) is applied to calculate intermolecular interaction energies in an extensive benchmark set consisting mainly of DNA base pairs and amino acid pairs, for which CCSD(T) complete basis set limit estimates are available (JSCH-2005 database). The three generalized gradient approximation (GGA) density functionals B-LYP, PBE and the new B97-D are tested together with the popular hybrid functional B3-LYP. The DFT-D interaction energies deviate on average by less than 1 kcal mol−1 or 10% from the reference values. In only six out of 161 cases, the deviation exceeds 2 kcal mol−1. With one exception, the few larger deviations occur for non-equilibrium structures extracted from experimental geometries. The largest absolute deviations are observed for pairs of oppositely charged amino acids which are, however, not significant on a relative basis due to the huge interaction energies >100 kcal mol−1 involved. The counterpoise (CP) correction for the basis set superposition error with the applied triple-zeta AO basis sets varies between 0 and −1 kcal mol−1 (<5% of the interaction energy in most cases) except for four complexes, where it is up to −1.4 kcal mol−1. It is thus suggested to skip the laborious calculation of the CP correction in DFT-D treatments with reasonable basis sets. The three dispersion corrected GGAs considered differ mainly for the interactions of the hydrogen-bonded DNA base pairs, which are systematically too small by 0.6 kcal mol−1 in case of B97-D, while for PBE-D they are too high by 1.5 kcal mol−1, and for B-LYP-D by 0.5 kcal mol−1. The all in all excellent results that have been obtained at affordable computational costs suggest the DFT-D method to be a routine tool for many applications in organic chemistry or biochemistry.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


