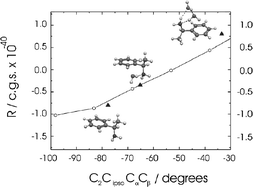
A computational (ab initio and molecular dynamics) and experimental exploration of the relative importance of molecular conformation and explicit solvent effects on the electronic circular dichroism (ECD) of chiral molecules, is presented. The exploration includes an assessment of the validity of angular correlation (sector) rules linking ECD to molecular conformation. It is based upon studies of 1-(R) phenylethanol (including its Raman optical activity spectrum), the corresponding ‘benchmark’ base, 1-(R)-phenylethylamine and its protonated cation; their hydrated clusters in the gas phase; and their non-polar and aqueous solutions. Emphasis is placed on the influence of specific, hydrogen bonded interactions with the aqueous solvent. The theoretical validity of the (otherwise empirical) sector rule in the neutral molecules and in their specifically hydrated clusters has been established—but with a reversal of the ‘historical’ sign convention. Protonation of the amine leads to a breakdown of the conventional sector rule but the change in its ECD intensity can still be related to the side chain dihedral angular dependence of its rotatory strength, computed ab initio for its explicitly hydrated clusters. Comparisons between ECD spectra measured in aqueous and in hydrocarbon solutions and the results of molecular dynamics calculations for aqueous solutions at 300 K, identify solvent induced structural change as the principal determinant of their relative ECD spectral intensities. Further links connecting the structures and conformations of chiral molecules and their explicitly solvated clusters in the gas phase, to their structures and conformational populations in solution can be expected through measurement, ab initio computation and analysis of their vibrational, ROA spectra.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


