
Simulated force-induced unfolding of α-helices: dependence of stretching stability on primary sequence
Abstract
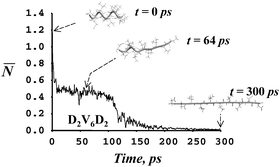
Some of the principles that determine a protein’s native fold can be probed with techniques for single-molecule manipulation. Yet, understanding the effects of an external force at atomic level still requires computer simulations. Here, we employ a novel protocol for steered molecular dynamics that allows for internal energy redistribution (and possibly, re-equilibration) while the molecule is subject to a mechanical perturbation. The approach is used to study how the stretching of α-helices is qualitatively affected by variations in primary sequence. Despite the simplifications introduces, our results indicate a trend whereby different amino acids can increase the resistance to mechanical unfolding depending on side chain polarity and the dynamics of side-chain internal torsions. Whereas the cooperative transition from α-helix to 310-helix and to a rod-like conformer prevails when stretching many sequences, we also find that the onset of the unfolding can be delayed by a range of alternative pathways which include events of helical refolding or long-lived intermediates with partial helical content.
 Please wait while we load your content...
Please wait while we load your content...

