
Thermal decomposition of O-benzyl ketoximes; role of reverse radical disproportionation
Abstract
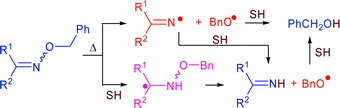
Thermolyses of seven dialkyl, two alkyl-aryl and two diaryl O-benzyl ketoxime ethers, R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOCH2Ph, have been examined in three hydrogen donor solvents: tetralin, 9,10-dihydrophenanthrene, and 9,10-dihydroanthracene. All the oxime ethers gave the products expected from homolytic scission of both the O–C bond (viz., R1R2CNOH and PhCH3) and N–O bond (viz., R1R2CNH and PhCH2OH). The yields of these products depended on which solvent was used and the rates of decomposition of the O-benzyl oxime ethers were greater in 9,10-dihydrophenanthrene and 9,10-dihydroanthracene than in tetralin. These results indicated that a reverse radical disproportionation reaction in which a hydrogen atom was transferred from the solvent to the oxime ether, followed by β-scission of the resultant aminoalkyl radical, must be important in the latter two solvents. Benzaldehyde was found to be an additional product from thermolyses conducted in tetralin. This, and other evidence, indicated that another induced decomposition mode involving abstraction of a benzylic hydrogen atom, followed by β-scission of the resulting benzyl radical, became important for some substrates. Participation by minor amounts of enamine tautomers of the oxime ethers was shown to be negligible by comparison of thermolysis data for the O-benzyloxime of bicyclo[3.3.1]nonan-9-one, which cannot give an enamine tautomer, with that of the O-benzyloxime of cyclohexanone.
NOCH2Ph, have been examined in three hydrogen donor solvents: tetralin, 9,10-dihydrophenanthrene, and 9,10-dihydroanthracene. All the oxime ethers gave the products expected from homolytic scission of both the O–C bond (viz., R1R2CNOH and PhCH3) and N–O bond (viz., R1R2CNH and PhCH2OH). The yields of these products depended on which solvent was used and the rates of decomposition of the O-benzyl oxime ethers were greater in 9,10-dihydrophenanthrene and 9,10-dihydroanthracene than in tetralin. These results indicated that a reverse radical disproportionation reaction in which a hydrogen atom was transferred from the solvent to the oxime ether, followed by β-scission of the resultant aminoalkyl radical, must be important in the latter two solvents. Benzaldehyde was found to be an additional product from thermolyses conducted in tetralin. This, and other evidence, indicated that another induced decomposition mode involving abstraction of a benzylic hydrogen atom, followed by β-scission of the resulting benzyl radical, became important for some substrates. Participation by minor amounts of enamine tautomers of the oxime ethers was shown to be negligible by comparison of thermolysis data for the O-benzyloxime of bicyclo[3.3.1]nonan-9-one, which cannot give an enamine tautomer, with that of the O-benzyloxime of cyclohexanone.
 Please wait while we load your content...
Please wait while we load your content...

