
Computational study of the spin-forbidden H2 oxidative addition to 16-electron Fe(0) complexes†
Abstract
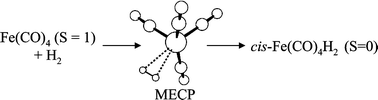
The spin-forbidden oxidative addition of H2 to Fe(CO)4, Fe(PH3)4, Fe(dpe)2 and Fe(dmpe)2 [dpe = H2PCH2CH2PH2, dmpe = (CH3)2PCH2CH2P(CH3)2] has been investigated by density functional theory using a modified B3PW91 functional. All 16-electron fragments are found to adopt a spin triplet ground state. The H2 addition involves a spin crossover in the reagents region of configurational space, at a significantly higher energy relative to the triplet dissociation asymptote and, for the case of Fe(CO)4·H2, even higher than the singlet dissociation asymptote. After crossing to the singlet surface, the addition proceeds directly to the classical cis-dihydride product. Only for the Fe(CO)4 was it possible to locate a stable energy minimum for the non-classical tautomer (dihydrogen complex), but the energy difference between this minimum and the tautomerisation transition state inverts when taking into account the zero-point energy correction. The geometries at the crossing points indicate a “side-on” approach of the H2 molecule to the metal for the Fe(CO)4, Fe(CO)2(PH3)2 and Fe(PH3)4 systems. These geometries are more reactants-like for the Fe(CO)4 system and more product-like for the Fe(PH3)4 system. The crossing point geometry for the Fe(dpe)2 system, on the other hand, is nearly C2-symmetric. The presence of an energy barrier on going from 3FeL4 + H2 to the crossing point is in agreement with the slow observed rates for addition of H2 to these unsaturated organometallic fragments.
 Please wait while we load your content...
Please wait while we load your content...

