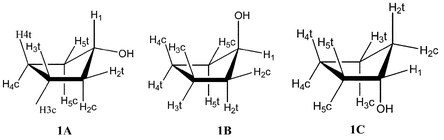
The conformations of cyclopentanol and cis- and trans-cyclopentane-1,2-diol have been studied by ab initio and molecular mechanics (MM) calculations and by the LIS technique, using Yb(fod)3 to obtain the induced shifts of all 1H and 13C nuclei in the molecule, together with complexation shifts obtained by the use of La(fod)3. The MM calculations gave two optimised geometries for cyclopentanol. These were envelope conformations with the hydroxyl group equatorial (1A) and axial (1B) at the flap of the envelope. In contrast Gaussian 98 at the B3LYP level with the 6-31G** basis set gave an optimised geometry (1C) which was an envelope conformation with the hydroxyl group in an axial position at the fold of the envelope. ΔE(1A − 1B)
= 0.47 kcal mol−1
(MM) and 0.93 kcal mol−1
(ab initio) and ΔE (1B − 1C)
= 0.15 kcal mol−1
(ab initio). The MM and ab initio calculations for cis-1,2-cyclopentanediol gave different envelope conformations (2A) and (2B), both with one equatorial and one axial hydroxyl group. For trans-1,2-cyclopentanediol both calculations gave the same geometries, an envelope conformation with two axial hydroxyls (3A) and a half chair conformer with diequatorial hydroxyls (3B). ΔE (3A − 3B)
= 2.9 kcal mol−1
( MM) and 0.70 kcal mol−1
(ab initio). The LIRAS4 model involving an sp3 hybridised oxygen atom with two symmetric lone pairs was used for these compounds. The calculated LIS for cyclopentanol gave poor agreement with the observed data for 1A, moderate agreement for 1B but good agreement for 1C. A LIS analysis combining 1B and 1C suggests that the population of 1C was >80% in CHCl3 solution. The ab initio calculations and the LIS analysis agree that the unsymmetric conformer 1C is the major form in solution. The similarity between this conformer of cyclopentanol and that of the furanose sugars suggests that the anomeric effect may be more fundamental than hitherto realised. In cis-cyclopentane-1,2-diol the observed data were in good agreement with the calculated LIS for both 2A and 2B. In trans-cyclopentane-1,2-diol the observed data were in good agreement with the calculated LIS for 3B but in poor agreement for 3A. The LIS allowed the assignment of the proton chemical shifts of the individual methylene protons in these molecules which had not been given previously.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
