
The structure, modelling and dynamics of hindered 5,6-diarylacenaphthenes
Abstract
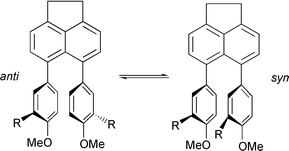
A series of 5,6-diarylacenaphthenes, 2–4, has been investigated using a variety of theoretical and experimental methods. The purpose of the investigation was to gain a thorough understanding of the dynamics of atropisomer interconversion in these molecules. Quantum chemical calculations were performed at different levels, including Hartree–Fock theory, density functional theory (B3LYP), and a semi-empirical method (AM1). Basis sets used ranged from STO-3G to 6-31+G*. The structures of energy minima and transition states for anti–syn interconversion were fully optimised. A geometrical comparison of the single-crystal X-ray structures of syn-2b, anti-2c and anti-2d was made with the results from the calculations, and revealed excellent agreement in most cases. Theoretical barriers to rotation were compared with those derived experimentally by
 Please wait while we load your content...
Please wait while we load your content...