
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced chemistry for the design of oligonucleotide conjugates and surfaces†
Antonina
Vigovskaya
ab,
Doris
Abt
bc,
Ishtiaq
Ahmed
b,
Christof M.
Niemeyer
b,
Christopher
Barner-Kowollik
*bc and
Ljiljana
Fruk
*ad
aDFG-Centre for Functional Nanostructures, Karlsruhe Institute of Technology (KIT), Wolfgang-Gaede-Str. 1a, 76131 Karlsruhe, Germany
bInstitut für Biologische Grenzflächen, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
dDepartment of Chemical Engineering and Biotechnology, University of Cambridge, New Museums Site, Pembroke Street, Cambridge, CB2 3RA, UK. E-mail: lf389@cam.ac.uk
First published on 1st December 2015
Abstract
A photocaged diene is introduced at the 5′-end of oligonucleotides using the H-phosphonate approach. The photoenol-functionalized DNA is subsequently employed for the conjugation to a protein and the spatially controlled immobilization onto surfaces using a light-induced Diels–Alder cycloaddition. Fully functional protein–DNA conjugates and patterned DNA surfaces are obtained under mild irradiation conditions.
Introduction
Nucleic acids are genetic information carriers, yet due to their structural properties have also found applications as structural elements for the design of nanotechnological constructs,1–3 used both in the new generation of functional materials4 or biosensing platforms.5–7 The range of the possible nucleic acid applications can be extended by introducing various chemical modifications to their structural elements either to prepare different conjugates8 or to immobilize DNA onto various surfaces.9–11 To date, a range of strategies has been employed to achieve these aims, including various procedures based on covalent binding12,13 as well as more specific methods such as cofactor reconstitution.14 In general, to achieve subsequent conjugation or immobilization, DNA needs to be first modified with the appropriate functional group and the most commonly employed strategy involves addition of such groups to the 5′- and 3′-termini of oligonucleotides as these have no affect on the Watson–Crick base pairing and the stability of the formed double helices,15 although the modified bases can also be employed for internal labelling of oligonucleotides.16,17 Functional groups are commonly introduced using standard phosphoramidite chemistry developed by Caruthers and co-workers18 using DNA synthesis on solid supports.19 A coupling step involving tetrazole induced activation results in exceptionally high yields and fast coupling reactions. However, the high reactivity is also associated with instability. Thus, phosphoramidites need to be handled carefully and exclusively under inert reaction conditions. Moreover, not all functional groups are compatible with phosphoramidite chemistry.20 To allow for the addition of specific functional groups, post-synthetic modification of DNA containing thiol or amine moieties is often employed such as the covalent attachment of different species via amide coupling or click chemistry procedures.21,22Recently, we have been particularly interested in exploring new photo-induced chemical strategies such as light triggered Diels–Alder cycloadditions23–26 to design nanoparticle bioconjugates27 or control the immobilization of various species onto different surfaces such as gold or glass.28,29 As such types of chemical reaction avoid the use of harsh conditions and as they are controlled by light, they are particularly interesting for surface modification and biointerface design, which can, in turn, be employed for the synthesis of bioinspired materials.30 Herein, we present the preparation of oligonucleotides modified with hydroxyl-o-quinodimethanes (photoenol, PE) and their use for both the preparation of DNA–protein conjugates and surface patterning (Scheme 1). To achieve that, a light triggered Diels–Alder conjugation, based on the [4+2] cycloaddition of a PE generated by the photoisomerisation of o-methylphenyl ketones or aldehydes, was applied to a range of different dienophiles such as maleimides.31–33 The photoenol mediated conjugation strategy requires no catalyst and takes place at ambient temperature. Moreover, it offers temporal and spatial control over the cycloaddition reaction as already shown in the case of biomolecule and polymer immobilization onto variable surfaces34 as well as the functionalization of nanoparticles29 and proteins.35
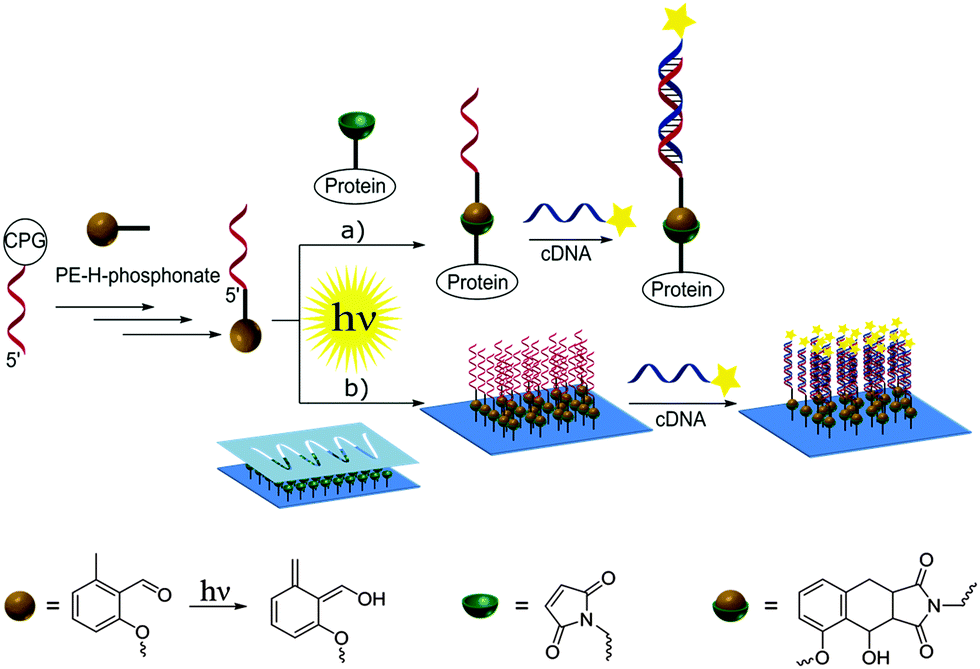 | ||
| Scheme 1 Introduction of the photoenol moiety into the DNA and subsequent light-induced conjugation to the protein (a) and immobilisation onto the silicon surface as well as hybridisation with the complementary DNA labelled with fluorophore (b). | ||
Experimental
Materials
Triethylene glycol (≥99%, Sigma-Aldrich), anhydrous N,N-dimethylformamide (DMF, ≥99.8%, Sigma-Aldrich), dichloromethane (DCM, ≥99.8%, VWR), 1-hydroxybenzotriazole hydrate (HOBt, ≥97%, Sigma Aldrich), N,N-dicyclohexylcarbodiimide (DCC, 99%, Sigma-Aldrich), N-hydroxysuccinimide (NHS, 98%, Sigma-Aldrich), methanol (MeOH, 99.8%, VWR), phosphorous acid (H3PO3, 99%, Sigma-Aldrich), anhydrous pyridine (99.8%, Sigma-Aldrich), trimethylacetyl chloride (99%, Sigma-Aldrich), acetonitrile (MeCN, 99.9%, VWR), anhydrous acetonitrile (MeCN, 99.8%, Sigma-Aldrich), triethylamine (NEt3, ≥99%, Sigma-Aldrich), 4-(N-maleimidomethyl)cyclohexane-1-carboxylic acid 3-sulfo-N-hydroxysuccinimide ester sodium salt (sulfo-SMCC, powder, Sigma-Aldrich), horseradish peroxidase (HRP, type VI-A, Sigma-Aldrich), water (MilliQ (MQ) system, Millipore), oxidizing solution (0.02 M iodine in THF/pyridine/water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1), Link Technologies), oligonucleotides DNA1 and DNA2 on solid support, cDNA1, 6-FAM-cDNA1, Cy®3-cDNA1 and Cy®3-DNA3 (Sigma-Aldrich, for sequences refer to Table S1, ESI†) were used as received. Protein marker, Precision Plus Protein™ Dual Xtra Standards, was purchased from Bio-Rad. DNA marker, O'Range Ruler 10 bp DNA Ladder, was purchased from ThermoFisher Scientific. Triethyl ammonium bicarbonate buffer (TEAB buffer, 2 M, pH 7.5) was prepared by bubbling of gaseous CO2 into 2 M aqueous solution of triethylamine until the pH of this solution reaches pH 7.5. 4-((2-formyl-3-methyl-phenoxy) methyl) benzoic acid241, 2-(2-(2-aminoethoxy)ethoxy)ethan-1-ol82 and N-(2-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)ethyl)-3-(2-(2-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethoxy)ethoxy)-ethoxy)propanamide36 (Do-TEG-Mal) were synthesized according to literature procedures. Gel filtration NAP5 and NAP10 columns and Vivaspin sample concentrators were purchased from GE Healthcare (Germany).
:2:1), Link Technologies), oligonucleotides DNA1 and DNA2 on solid support, cDNA1, 6-FAM-cDNA1, Cy®3-cDNA1 and Cy®3-DNA3 (Sigma-Aldrich, for sequences refer to Table S1, ESI†) were used as received. Protein marker, Precision Plus Protein™ Dual Xtra Standards, was purchased from Bio-Rad. DNA marker, O'Range Ruler 10 bp DNA Ladder, was purchased from ThermoFisher Scientific. Triethyl ammonium bicarbonate buffer (TEAB buffer, 2 M, pH 7.5) was prepared by bubbling of gaseous CO2 into 2 M aqueous solution of triethylamine until the pH of this solution reaches pH 7.5. 4-((2-formyl-3-methyl-phenoxy) methyl) benzoic acid241, 2-(2-(2-aminoethoxy)ethoxy)ethan-1-ol82 and N-(2-(2,2-dimethylbenzo[d][1,3]dioxol-5-yl)ethyl)-3-(2-(2-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethoxy)ethoxy)-ethoxy)propanamide36 (Do-TEG-Mal) were synthesized according to literature procedures. Gel filtration NAP5 and NAP10 columns and Vivaspin sample concentrators were purchased from GE Healthcare (Germany).
Characterisation
1H, 13C and 31P NMR spectroscopy was performed using Bruker AM 250, Bruker AM 300 or Bruker AM 400 spectrometer at 250 MHz or 400 MHz respectively. Samples were dissolved in CDCl3 or CD3CN. The δ-scale is referenced to tetramethylsilane as the internal standard. Fast Atom Bombardment Mass Spectrometry (FAB-MS): the mass spectra were measured using Finnigan MAT90 mass spectrometer. Fast protein liquid chromatography (FPLC) was performed using an Äkta explorer system, which was connected to a MonoQ 5/50 GL anion exchange column (GE Healthcare, Germany). Used purification conditions: buffer A: 20 mM Tris; buffer B: 20 mM Tris, 1 M NaCl; gradient: linear increase of buffer B to 100% in 30 minutes, flowrate: 1 mL min−1, UV/Vis detection at 260 nm and 403 nm. High-performance liquid chromatography (HPLC) was performed using HPLC system 1200 series of Agilent Technologies connected to a Zorbax Eclipse XDB-C18 column (dimensions 4.6 × 150 mm) from Agilent. The following purification conditions were used: (A) 0.1 M NH4OAc, (B) acetonitrile, gradient 0–100% B over 40 min, flowrate 1 mL min−1, UV/Vis detection at 260 nm and 280 nm. Purification was verified by MS (MALDI-TOF) on a Autoflex III SmartBeam™ (Nd:YAG laser (355 nm) with a repetition rate of 200 Hz) spectrometer from Bruker Daltonics in the linear negative mode; used matrix: a 9:1 mixture of saturated 3-hydroxypicolinic acid and 0.44 M diammonium hydrogen citrate. Polyacrylamide gel electrophoresis (PAGE) characterization was performed by using a Mini-Protean® Tetra System, which was connected to a PowerPac™ voltage source (BioRad, Germany). Fourier transform infrared spectra (FTIR) were recorded using Bruker ALPHA and performed in attenuated total reflection (ATR) mode. Time-of-flight secondary ion mass spectrometry (ToF-SIMS) was conducted with a TOF·SIMS5 instrument (ION-TOF GmbH, Münster, Germany), equipped with a Bi cluster liquid metal primary ion source and a non-linear time-of-flight analyzer. The Bi source was operated in the bunched mode providing 0.7 ns Bi3+ ion pulses at 25 keV energy and a lateral resolution of approx. 4 μm. The short pulse length allowed high mass resolution to analyze the complex mass spectra of the immobilized organic layers. Images larger than the maximum deflection range of the primary ion gun of 500 × 500 μm2 were obtained using the manipulator stage scan mode. Primary ion doses were kept below 1011 ions m−2 (static SIMS limit). Spectra were calibrated on the CH−, CH2−, CH3−, or on the C+, CH+, CH2+, and CH3+ peaks. Fluorescence microscopy was performed using an Axiovert 200 M (Carl Zeiss) inverted microscope with Plan-Neofluar objective (magnification/numeric aperture 5×/0.16).
Synthesis of PE H-phosphonate 4
:1, 1:1, 1:2 (v/v), MeOH), Rf = 0.14 (EtOAc), to give the title compound 3 as yellow solid (yield: 69.8 mg, 47%) (refer to ESI,† for IR, NMR and MS data, Fig. S1 and S2).
:1, v/v) (33.4 mg, 0.36 M) under argon atmosphere and added to the CPG. Pivaloyl chloride (250 μL, 100 mM in dry CH3CN/pyridine (1:1, v/v)) was added and the reaction mixture was stirred at 39 °C for 16 hours. The reaction solution was removed by centrifugation and the solid support washed with CH3CN (1 mL × 3). After treatment with standard DNA synthesis oxidizing solution for 3 minutes the CPG was again washed with CH3CN (1 mL × 3). The PE-modified sequence was deprotected and cleaved from the solid support by incubation with 500 μL 25% ammonia solution at 55 °C for 5 hours. The supernatant was collected and the solvents removed in vacuo. Photoenol modified oligonucleotide was dissolved in water and purified by reversed phase HPLC using C18 column (eluent A: 0.100 M ammonium acetate, eluent B: CH3CN). The main fractions were collected, concentrated in vacuo and characterized by MALDI-TOF and PAGE (Fig. S4–S6, ESI†).
General procedure for the photo-induced reactions
The samples to be irradiated were crimped air-tight in headspace vials (20 mm, VWR, Germany) using SBR seals (VWR, Germany) with PTFE inner liner. The photoreactions were performed in a custom-built photoreactor (Fig. S7, ESI†), consisting of a metal disk which revolves at a distance of 40–50 mm around a compact low-pressure fluorescent lamp with λmax = 320 nm ± 30 nm (36 W, Arimed B6, Cosmedico GmbH, Germany) (Fig. S8, ESI†). For the spatially controlled surface immobilization of PE-DNA1, maleimide functionalized surfaces were mounted into sample holders with a shaddow mask before being immersed with the reaction solution.Photoreaction between PE-DNA1 and Do-TEG-Mal
The mixture of 2.00 nmol PE-DNA1 and 2.00 nmol Do-TEG-Mal in 1.00 mL H2O/CH3CN (1:1, v/v) was placed into the headspace vial, which was crimped air-tight as described above, degassed by purging with nitrogen for 15 minutes and subsequently irradiated for 16 hours in the photoreactor at λmax = 320 nm. After the irradiation solvents were removed under reduced pressure, the residue was redissolved in water and purified by reversed phase HPLC using C18 column (eluent A: 0.100 M ammonium acetate, eluent B: CH3CN). The collected fractions were characterized by MALDI-TOF and native PAGE (Fig. S9 and S10, ESI†).
Photoreaction between PE-DNA1 and HRP-Mal
First HRP was functionalized with a maleimide group in the coupling reaction with sulfo-SMCC. Sulfo-SMCC (2.00 mg, 4.58 μmol) was dissolved in 100 μL DMF and added to 200 μL HRP solution (166 μM in PBS buffer, pH 6.0) and incubated for 1 hour at ambient temperature. The excess sulfo-SMCC was removed by filtration of the reaction mixture through NAP5 and NAP10 columns. The buffer was exchanged using PBS buffer pH 7.1 as eluent. For the photoreaction 2.00 nmol of PE-DNA1 and 2.20 nmol of fresh prepared HRP-Mal were dissolved in 500 μL of PBS/CH3CN solution (3:2, v/v) and placed into the headspace vial, which was crimped air-tight as described above, degassed by purging with nitrogen for 15 minutes and subsequently irradiated for 16 hours in the photoreactor at λmax = 320 nm. The reaction mixture was filtered through NAP5 and NAP10 columns to exchange the buffer to 20 mM Tris/HCl pH 8.3, concentrated by using 5 kDa Vivaspin and purified by anion exchange chromatography using MonoQ 5/50 GL column (buffer A: 20 mM Tris, buffer B: 20 mM Tris, 1 M NaCl; gradient: linear increase of buffer B to 100% in 30 minutes, flowrate: 1 mL min−1). The concentration and buffer exchange to PBS of the collected fractions were carried out by using 5 kDa Vivaspin, and the characterisation of product fraction was performed by native PAGE.
Spatially controlled surface functionalisation with PE-DNA1 and subsequent hybridization with complementary DNA labelled with Cy®3 dye (Cy®3-cDNA1)
In a headspace vial (Pyrex, diameter 20 mm) 7 nmol PE-DNA1 were dissolved in 4 mL of PBS/ACN (1:1) mixture. Maleimide functionalized silicon surface mounted into the sample holder with mask was placed into the PE-DNA1 solution. The vial was crimped air-tight using SBR seals with PTFE inner liner. The solution was deoxygenated by purging with nitrogen for 15 min and subsequently irradiated for 5 h in the photoreactor. After irradiation, the mask was removed. The surface was rinsed with MQ water and sonicated 10 seconds in water to remove any possibly physisorbed material. The surface was subsequently washed with PBS buffer, MQ water, DMF, DMSO, again with MQ water and finally dried under a nitrogen stream. The surface was then analyzed by ToF-SIMS. For the control experiment the same procedure as described above was applied for the non-modified DNA1. ToF-SIMS images show no fragments corresponding to DNA1 (refer to Fig. S16, ESI†). For the hybridization experiments, the surface with immobilized PE-DNA1 was covered with a 50 μM solution of complementary DNA labeled with Cy®3 dye (λExc = 550 nm, λEm = 570 nm) in TETBS buffer (pH = 7.5) and incubated overnight in the dark at ambient temperature. The surface was then washed several times with MQ water, TETBS buffer and again with MQ water, dried under a nitrogen stream and analyzed by fluorescence microscopy. For the control experiment, the surface with immobilized PE-DNA1 was covered with non-complementary DNA labeled with Cy®3 dye using the same procedure as described above. No fluorescence pattern corresponding to the mask features could be detected (refer to Fig. S17, ESI†).
Results and discussion
Previously, we have used amide coupling of photoenol carboxylic acid to amine modified DNA to prepare photoenol-functional DNA.35 However, such coupling reactions usually result in minute amounts of functional DNA and involve several steps, which can be circumvented by direct coupling of the PE to the 5′-end of the oligonucleotide during the solid-phase synthesis. To achieve a direct coupling, we decided to employ an H-phosphonate-based strategy instead of commonly used phosphoramidites, as H-phosphonates are easier to handle and more resistant towards oxidation than other P(III) compounds, due to the absence of the lone electron pair located on the phosphorus atom (a consequence of the tetrahedral geometry of H-phosphonates, which contain a phosphoryl group (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and a hydrogen atom bound to the phosphorus centre).38 First reports on the use of H-phosphonate in nucleotide chemistry originated in the 1950s,39 yet their use in solid phase synthesis was only demonstrated three decades later by Garegg et al.40 as well as Froehler and Matteucci.41
O) and a hydrogen atom bound to the phosphorus centre).38 First reports on the use of H-phosphonate in nucleotide chemistry originated in the 1950s,39 yet their use in solid phase synthesis was only demonstrated three decades later by Garegg et al.40 as well as Froehler and Matteucci.41
The PE functional H-phosphonate 4 is prepared as shown in Fig. 1. In the first step, an OH group containing precursor is required to enable the subsequent reaction with P(III) compounds (e.g., H3PO4 or PCl3) in order to afford an H-phosphonate derivative. Therefore, we have first prepared PE 3 containing a PE function and a three membered glycol unit to allow for more steric freedom for the following ligation. Subsequently, the PE H-phosphonate 4 was directly obtained from PE 3 using the modified procedure described by Dougan and co-workers.20 The PE modified oligonucleotide sequences DNA1 and DNA2 were obtained by direct coupling to commercially available CPG-bound oligonucleotides using the PE-H-phosphonate 4 and pivaloyl chloride as activator. The products, PE-DNA1 and PE-DNA2, were purified using reversed phase HPLC and identified by MALDI-TOF mass spectrometry (Fig. S4 and S5, ESI†). Gel electrophoresis was additionally used for the characterisation of both PE-DNAs (Fig. S6, ESI†).
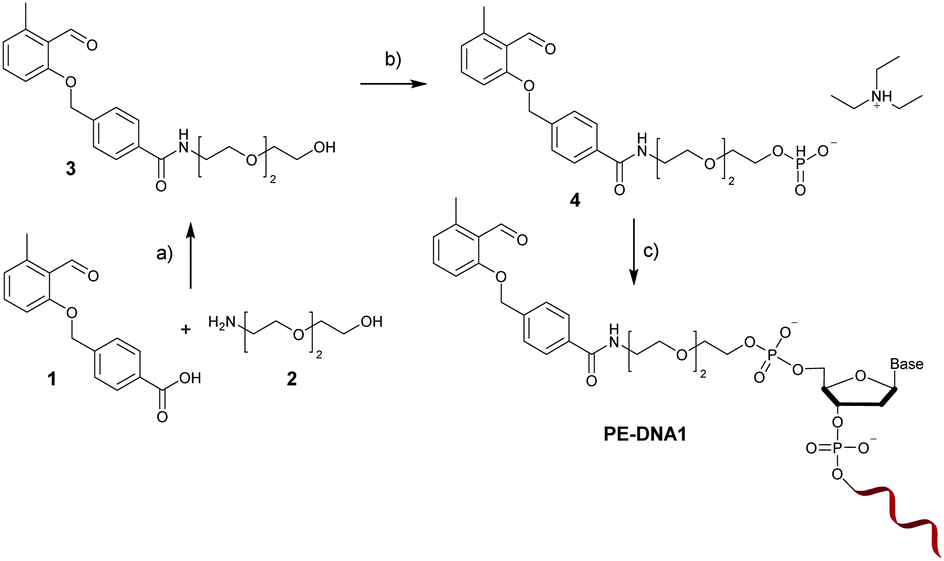 | ||
| Fig. 1 Synthesis of the photoenol containing oligonucleotide PE-DNA1: (a) HOBt, DCC, NHS, DMF, r.t., 48 h, 47%; (b) H3PO3, pivaloyl chloride, pyridine, r.t., 2 h, TEAB buffer; (c) solid phase synthesis. | ||
To allow for the light-induced cycloaddition, irradiation of PE oligonucleotides with a UVA lamp (λmax = 320 nm) in the presence of maleimide containing molecules of interest is needed. Previous reports have shown that the UV irradiation with the same wavelength of non-modified DNA in PBS/CH3CN (1:1) had no destructive effect on the functionality of the oligonucleotide.35 In order to investigate the influence of the irradiation on the PE modified oligonucleotide, a proof of concept reaction was initially performed by irradiating PE-DNA1 in the presence of the maleimide containing molecule Do-TEG-Mal (Fig. 2). The HPLC purification and subsequent analysis by gel electrophoresis and MALDI-TOF showed that the reaction was successful and the desired product was obtained (Fig. S9, ESI†). In addition, PE-DNA2 with a different sequence was used for the reaction, indicating that the light induced photoenol reaction is not sequence specific (Fig. S10, ESI†). When the reaction is performed using non-modified DNA, no product was obtained (Fig. S11, ESI†).
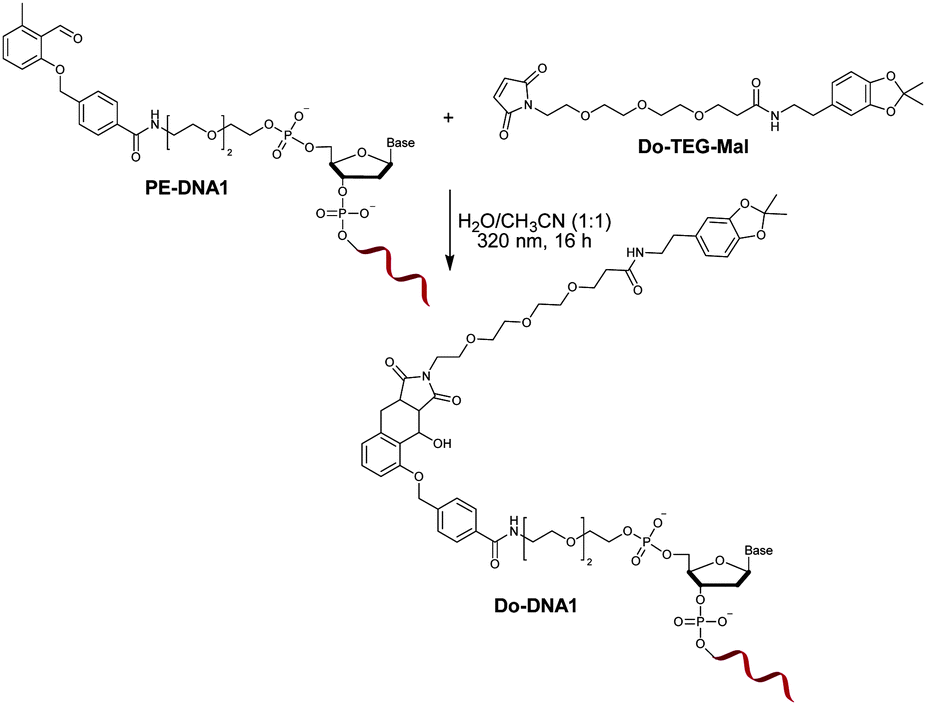 | ||
| Fig. 2 Light-induced conjugation of PE-DNA1 and Do-TEG-Mal. | ||
Furthermore, a control reaction was carried out by irradiating PE-DNA1 alone. Here, some signals with shorter retention time and lower absorption intensity were detected during HPLC (refer to Fig. S12, ESI†), indicating that there are possible side reactions of the photoenol moiety and the nucleobases upon UV irradiation, most probably with available amine groups. However, photoreactions in the presence of the dienophile Do-TEG-Mal revealed the desired Do-DNA1 cycloadduct as the main product (refer to Fig. S12, ESI†) evidenced by HPLC analysis as well as by MALDI-TOF (refer to Fig. S9, ESI†). Furthermore, an additional control experiment was carried out in which PE-DNA1 and Do-TEG-Mal were incubated overnight in the absence of light, showing that there is no significant amount of product or side products formed (refer to Fig. S12, ESI†).
It should be noted that overnight irradiation was performed to ensure the maximum conversion of the starting material to the cycloadduct in aqueous solution, as protic solvents favor the reconversion of dienol species to ketone/aldehyde tautomers.24,42 As reported by Tchir and Porter, five transients have been observed via laser flash photolysis of 2,4-dimethylbenzophenone in cyclohexane or ethanol.43 The dienol intermediate exists as a mixture of two isomers (E- and Z-isomer). The Z isomer decays much faster than the E isomer. Measured lifetimes of the dienols in cyclohexane were 4 s for the Z- and 250 s for the E-dienol. Arnold et al. described a similar photochemical process for 2-methylbenzaldehyde, where the E- and Z-dienols are the expected initial products.44 The lifetimes of these dienols are not reported, yet the authors claim that the cycloaddition reaction with a dienophile is efficient and depends on the rate of dienol formation.
To demonstrate that the photo-induced cycloaddition of PE modified oligonucleotide can further be employed in more complex applications, protein–DNA conjugates using maleimide containing horseradish peroxidase (HRP-Mal) were prepared. HRP is a biotechnologically very important protein and one of the most potent peroxidases.45 As proteins are, in general, sensitive to UV light, the effect of prolonged irradiation was first assessed by irradiating native HRP with λmax = 320 nm light overnight. SDS-PAGE analysis showed neither a difference in electrophoretic mobility between irradiated and non-irradiated HRP nor additional fragments for irradiated samples (Fig. S15, ESI†), indicating that there is no significant structural damage of the protein under the employed irradiation conditions. For the preparation of protein–DNA conjugates, the mixture of HRP-Mal and PE-DNA1 was irradiated in PBS/CH3CN (3:2) overnight (Fig. 3a), purified by fast protein liquid chromatography (Fig. 3b) and analyzed by gel electrophoresis (Fig. 3c). The gel characterization showed that the peak F1 represents the non-modified HRP-Mal. The peak F2 was assigned to the HRP–DNA1 photoconjugate, as it shows the higher electrophoretic mobility in comparison to HRP and HRP-Mal due to the increased negative charge after the coupling to PE-DNA1. In addition, the presence of the oligonucleotide in the HRP–DNA1 fraction was evidenced by SYBR Gold staining. Fraction F3 represents the non-reacted PE-DNA1. In order to explore if any non-specific side reactions take place between modified and unmodified HRP and DNA1, a set of control reactions was performed (Fig. S13, ESI†). In case of the reaction between native HRP and PE-DNA1 under irradiation, a small amount of conjugate could be detected in the FPLC chromatogram, indicating the occurrence of a reaction of the photoenol moiety with the protein – possibly with free lysine groups. However, the significant amount of the conjugate obtained in the photoreaction of HRP-Mal and PE-DNA1 shows that the photoenol-mediated cycloaddition is preferred (Fig. S14, ESI†). The selectivity of the photoreaction was additionally confirmed by the absence of conjugate after the incubation of HRP-Mal and PE-DNA1 in the dark as well as in case of irradiation of PE-DNA1 with HRP or with HRP-Mal (Fig. S13, ESI†).
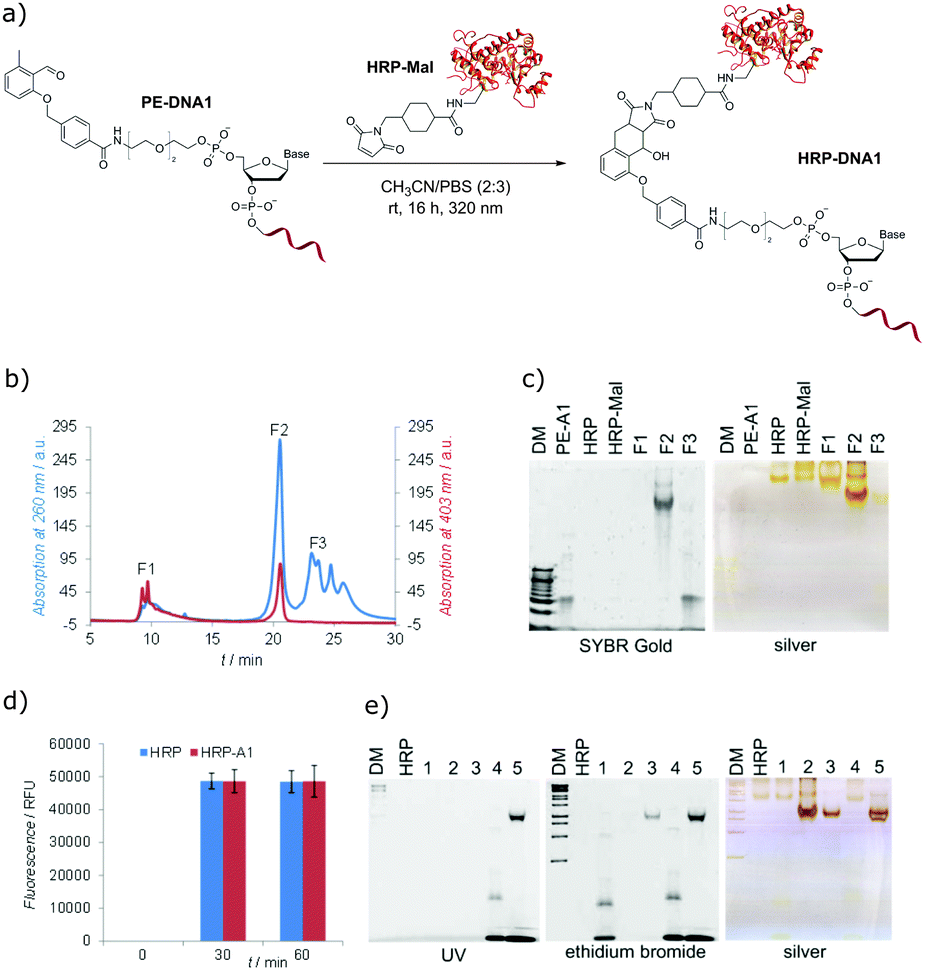 | ||
| Fig. 3 (a) Photo-triggered conjugation of PE-DNA1 and maleimide functionalized HRP (HRP-Mal); (b) FPLC chromatogram of the HRP–DNA1 purification; (c) native PAGE characterization of the FPLC fractions F1, F2 and F3; oligonucleotide/protein visualization by SYBR Gold/silver staining; DM: 10 bp DNA-marker; (d) fluorescence HRP activity assay; (e) native PAGE analysis of hybridization of HRP–DNA1 with complementary strands cDNA1 and 6-FAM-cDNA1, DM: 1 kb DNA-marker, HRP: native HRP; (1) negative control (HRP + DNA1 + cDNA1 or (4) HRP + DNA1 + 6-FAM-cDNA1), (2) HRP–DNA1 conjugate, (3) HRP–DNA1 + cDNA1, (5) HRP–DNA1 + 6-FAM-cDNA1; visualization: UV irradiation, ethidium bromide and silver staining. | ||
In order to investigate if the inherent functions of HRP and DNA are preserved in the HRP–DNA1 conjugate, the peroxidase activity of HRP and the hybridization ability of DNA were tested. The Amplex®Red assay was used to assess the peroxidase activity of the HRP–DNA1 conjugate, employing the conversion of non-fluorescent substrate Amplex Red into fluorescent resorufin in the presence of peroxidase enzymes and H2O2 (Scheme S2, ESI†). Fluorescence measurements (Fig. 3d) showed similar results for native HRP and HRP–DNA1 conjugate indicating the high peroxidase activity of the conjugate. For the hybridization ability test, the HRP–DNA1 conjugate was incubated with a 10-fold excess of cDNA1 and 6-FAM dye labelled cDNA1. As shown in Fig. 3e, hybridization success was confirmed by detection of 6-FAM fluorescence after UV irradiation, ethidium bromide and silver staining. Further, as negative controls, native HRP was incubated with DNA1, cDNA1 and with 6-FAM-cDNA1 and no HRP mobility shifts or fluorescence bands indicating the hybridization were observed after the ethidium bromide staining and UV irradiation.
The ability of 5′-photoenol-modified oligonucleotides to undergo a light-induced cycloaddition reaction with maleimide functionalized molecules in solution prompted us to employ it for the temporally and spatially controlled patterning of oligonucleotides and the design of functional material-biomolecule interfaces. Thus, we initially functionalized a silicon surface with maleimide groups using a previously reported procedure.46 The spatial control was achieved by utilizing a shadow mask with wave patterns (Fig. 4a).47 The maleimide functionalized silicon wafer, covered with a shadow mask, was irradiated in a solution of PE-DNA1 in PBS/CH3CN (1:1) for 5 hours. The control experiment was performed via the irradiation of non-modified DNA1 under the same reaction conditions. One issue that may arise upon surface modification is the formation of a potential self-adduct between two identical enol species, decreasing the efficiency of the DNA immobilisation. However, as reported by Arnold et al., the dimer formation between two 2-methylbenzaldehyde molecules could be detected at relatively high concentration (ca. 0.1 M) conditions.44 The dimerisation reaction in more diluted solutions (0.003 M) was found to be, however, disfavored. In our current study, solutions with less than 4 μM concentrations of enol species were employed, therefore the dimerisation reaction is not expected. In any case, even if dimerization has occurred, the dimers will not be attached to the surface and are washed off.
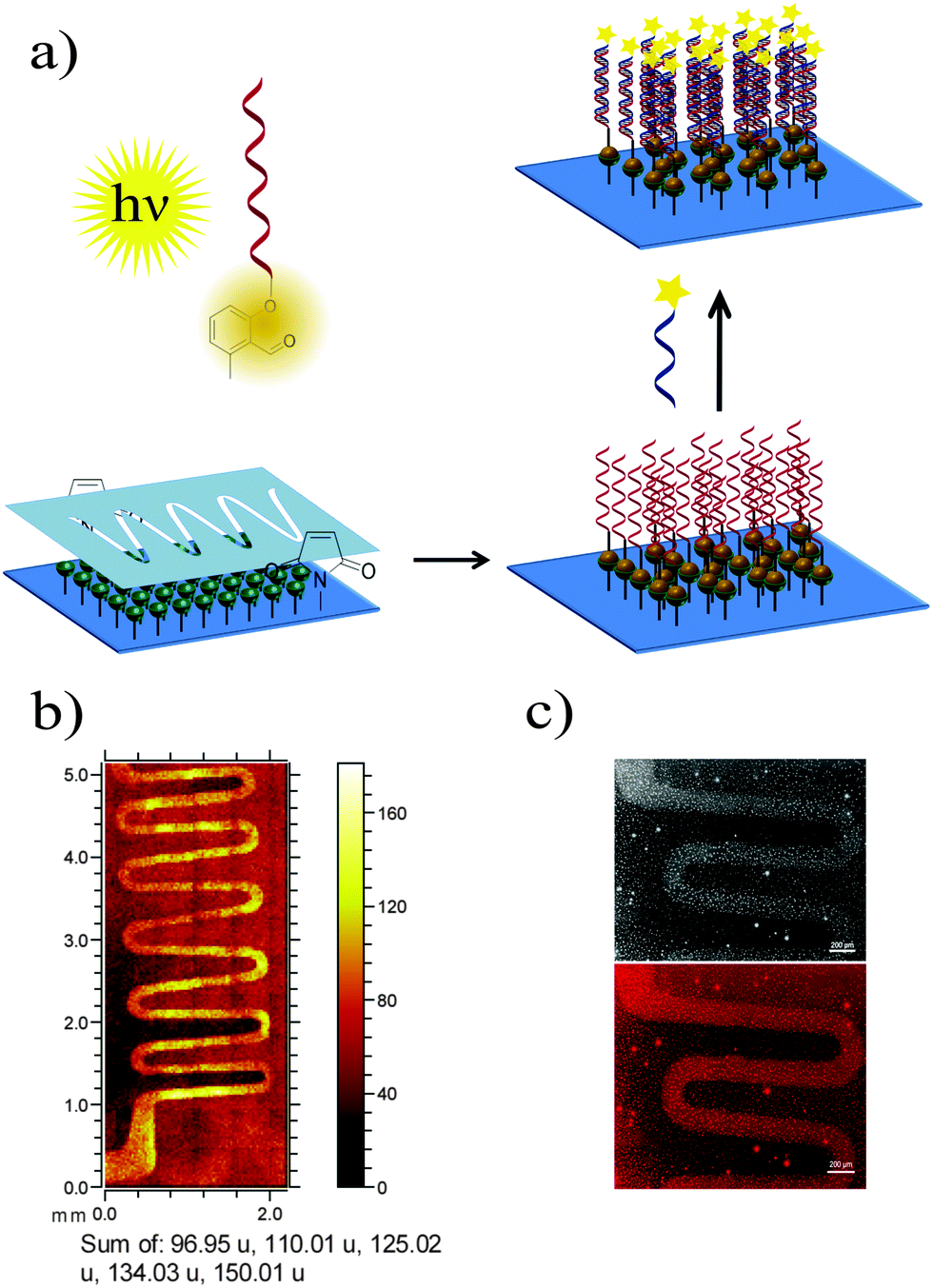 | ||
| Fig. 4 (a) Light-induced DNA immobilization with spatial resolution; (b) ToF-SIMS imaging of photo-patterned PE-DNA1 derived from the sum of the signals detected at 96.95 (H2PO4−), 150.01 (G−), 125.02 (T−), 134.03 (A−) and 110.01 (C−) assigned to fragments of the corresponding oligonucleotides; (c) fluorescence microscopy of hybridized cDNA1-Cy®3 on the surface patterned with PE-DNA1 (top: grey scale image, bottom: Cy®3 fluorescence image). | ||
The surfaces patterned with oligonucleotides were analyzed by time-of-flight secondary ion mass spectrometry (ToF-SIMS). Fig. 4b depicts the ToF-SIMS image resulting from the sum of characteristic species assigned to nucleic acids of DNA (m/z = 96.95 (H2PO4−), 150.01 (G−, guanine), 125.02 (T−, thymine), 134.03 (A−, adenine) and m/z = 110.01 (C−, cytosine))48 immobilized in a patterned fashion following the mask's features.
Once it had been shown that PE-DNA1 was successfully immobilized, we assessed if the surface bound DNA1 remained functional after irradiation. Thus, surfaces patterned with DNA were incubated with complementary oligonucleotide cDNA1 labelled with a commercially available fluorophore Cy®3 (λExc = 550 nm, λEm = 570 nm). The fluorescence detection of hybridized oligonucleotides was performed by fluorescence microscopy and shows a clear fluorescent pattern in the areas with immobilized DNA1 (Fig. 4c). The overnight hybridization leads to some limited aggregation of the cDNA1-Cy®3 and a subsequent adsorption of the aggregates onto the surfaces. The aggregates are visible as very small circular deposits in the non-irradiated areas of the array. Further optimization of the hybridization patterns is required. However, a clear fluorescent pattern was visible indicating successful DNA immobilization and subsequent hybridization, therefore allowing for future immobilisation of various species through DNA directed immobilization.
Conclusions
We have introduced a facile route to prepare 5′-photoenol-modified oligonucleotides via an H-phosphonate solid phase coupling method. The approach has proven to be a useful tool for oligonucleotide modification with a photocaged diene species which can further react with dienophiles such as functional maleimide using a light-induced [4+2] cycloaddition reaction. Employing photo-triggered reactions both protein–DNA (HRP–DNA) conjugates and patterned DNA surfaces were prepared. We believe that the introduced methodology can be readily expanded further for the controlled assembly of oligonucleotides onto structured surfaces to create, e.g., catalytic centres within artificial enzymes. Our further efforts are focused on designing a library of different photoenol species, which in the future will enable photoorthogonal DNA assembly.Acknowledgements
The current study was supported by DFG-CFN grant A 5.7. C. B.-K. and C. M. N. acknowledge continued support from the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz programme BioInterfaces in Technology and Medicine (BIFTM). A. V. thanks Annette Hochgesand (KIT) for the MALDI-TOF measurements.References
- Y. C. Hung, D. M. Bauer, I. Ahmed and L. Fruk, Methods, 2014, 67, 105–115 CrossRef CAS PubMed.
- O. I. Wilner and I. Willner, Chem. Rev., 2012, 112, 2528–2556 CrossRef CAS PubMed.
- C. Geary, P. W. Rothemund and E. S. Andersen, Science, 2014, 345, 799–804 CrossRef CAS PubMed.
- A. Kuzuya and M. Komiyama, Nanoscale, 2010, 2, 310–322 RSC.
- B. Hotzer, I. L. Medintz and N. Hildebrandt, Small, 2012, 8, 2297–2326 CrossRef PubMed.
- D. G. Thompson, A. Enright, K. Faulds, W. E. Smith and D. Graham, Anal. Chem., 2008, 80, 2805–2810 CrossRef CAS PubMed.
- C. S. Thaxton, R. Elghanian, A. D. Thomas, S. I. Stoeva, J. S. Lee, N. D. Smith, A. J. Schaeffer, H. Klocker, W. Horninger, G. Bartsch and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18437–18442 CrossRef CAS PubMed.
- D. M. Bauer, I. Ahmed, A. Vigovskaya and L. Fruk, Bioconjugate Chem., 2013, 24, 1094–1101 CrossRef CAS PubMed.
- G. Shtenberg, N. Massad-Ivanir, O. Moscovitz, S. Engin, M. Sharon, L. Fruk and E. Segal, Anal. Chem., 2013, 85, 1951–1956 CrossRef CAS PubMed.
- G. Shtenberg, N. Massad-Ivanir, S. Engin, M. Sharon, L. Fruk and E. Segal, Nanoscale Res. Lett., 2012, 7, 443 CrossRef PubMed.
- H. Schroeder, M. Adler, K. Gerigk, B. Muller-Chorus, F. Gotz and C. M. Niemeyer, Anal. Chem., 2009, 81, 1275–1279 CrossRef CAS PubMed.
- L. Fruk, J. Muller, G. Weber, A. Narvaez, E. Dominguez and C. M. Niemeyer, Chem. – Eur. J., 2007, 13, 5223–5231 CrossRef CAS PubMed.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200–1216 CrossRef CAS PubMed.
- L. Fruk and C. M. Niemeyer, Angew. Chem., Int. Ed., 2005, 44, 2603–2606 CrossRef CAS PubMed.
- G. N. Grimm, A. S. Boutorine and C. Helene, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 1943–1965 CAS.
- L. Fruk, A. Grondin, W. E. Smith and D. Graham, Chem. Commun., 2002, 2100–2101 RSC.
- I. K. Astakhova and J. Wengel, Chem. – Eur. J., 2013, 19, 1112–1122 CrossRef CAS PubMed.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- S. Roy and M. Caruthers, Molecules, 2013, 18, 14268–14284 CrossRef PubMed.
- J. A. Dougan, A. K. Reid and D. Graham, Tetrahedron Lett., 2010, 51, 5787–5790 CrossRef CAS.
- P. M. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442–3444 CrossRef CAS PubMed.
- M. Shelbourne, T. Brown, Jr., A. H. El-Sagheer and T. Brown, Chem. Commun., 2012, 48, 11184–11186 RSC.
- K. K. Oehlenschlaeger, J. O. Mueller, N. B. Heine, M. Glassner, N. K. Guimard, G. Delaittre, F. G. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2013, 52, 762–766 CrossRef CAS PubMed.
- T. Pauloehrl, G. Delaittre, V. Winkler, A. Welle, M. Bruns, H. G. Borner, A. M. Greiner, M. Bastmeyer and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2012, 51, 1071–1074 CrossRef CAS PubMed.
- A. S. Quick, H. Rothfuss, A. Welle, B. Richter, J. Fischer, M. Wegener and C. Barner-Kowollik, Adv. Funct. Mater., 2014, 24, 3571–3580 CrossRef CAS.
- K. Hiltebrandt, T. Pauloehrl, J. P. Blinco, K. Linkert, H. G. Borner and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2015, 54, 2838–2843 CrossRef CAS PubMed.
- L. Stolzer, A. Vigovskaya, C. Barner-Kowollik and L. Fruk, Chem. – Eur. J., 2015, 21, 14309–14313 CrossRef CAS PubMed.
- L. Stolzer, A. S. Quick, D. Abt, A. Welle, D. Naumenko, M. Lazzarino, M. Wegener, C. Barner-Kowollik and L. Fruk, Chem. Commun., 2015, 51, 3363–3366 RSC.
- L. Stolzer, I. Ahmed, C. Rodriguez-Emmenegger, V. Trouillet, P. Bockstaller, C. Barner-Kowollik and L. Fruk, Chem. Commun., 2014, 50, 4430–4433 RSC.
- C. M. Preuss, T. Tischer, C. Rodriguez-Emmenegger, M. M. Zieger, M. Bruns, A. S. Goldmann and C. Barner-Kowollik, J. Mater. Chem. B, 2014, 2, 36–40 RSC.
- J. L. Charlton and M. M. Alauddin, Tetrahedron, 1987, 43, 2873–2889 CrossRef CAS.
- S. M. Mellows and P. G. Sammes, J. Chem. Soc. D, 1971, 21–22 RSC.
- T. Gruendling, K. K. Oehlenschlaeger, E. Frick, M. Glassner, C. Schmid and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 807–812 CrossRef CAS PubMed.
- G. Delaittre, A. S. Goldmann, J. O. Mueller and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2015, 54, 11388–11403 CrossRef CAS PubMed.
- D. M. Bauer, A. Rogge, L. Stolzer, C. Barner-Kowollik and L. Fruk, Chem. Commun., 2013, 49, 8626–8628 RSC.
- C. Chen, I. Ahmed and L. Fruk, Nanoscale, 2013, 5, 11610–11614 RSC.
- T. D. Schladt, K. Schneider, M. I. Shukoor, F. Natalio, H. Bauer, M. N. Tahir, S. Weber, L. M. Schreiber, H. C. Schroder, W. E. G. Muller and W. Tremel, J. Mater. Chem., 2010, 20, 8297–8304 RSC.
- A. Kraszewski and J. Stawinski, Pure Appl. Chem., 2007, 79, 2217–2227 CrossRef CAS.
- N. S. Corby, G. W. Kenner and A. R. Todd, J. Chem. Soc., 1952, 3669–3675 RSC.
- P. J. Garegg, I. Lindh, T. Regberg, J. Stawinski, R. Stromberg and C. Henrichson, Tetrahedron Lett., 1986, 27, 4051–4054 CrossRef CAS.
- B. C. Froehler and M. D. Matteucci, Tetrahedron Lett., 1986, 27, 469–472 CrossRef CAS.
- P. G. Sammes, Tetrahedron, 1976, 32, 405–422 CrossRef CAS.
- G. Porter and M. F. Tchir, J. Chem. Soc. D, 1970, 1372–1373 RSC.
- B. J. Arnold, S. M. Mellows, P. G. Sammes and T. W. Wallace, J. Chem. Soc., Perkin Trans. 1, 1974, 401–409 RSC.
- F. W. Krainer and A. Glieder, Appl. Microbiol. Biotechnol., 2015, 99, 1611–1625 CrossRef CAS PubMed.
- B. Yameen, C. Rodriguez-Emmenegger, C. M. Preuss, O. Pop-Georgievski, E. Verveniotis, V. Trouillet, B. Rezek and C. Barner-Kowollik, Chem. Commun., 2013, 49, 8623–8625 RSC.
- T. Tischer, T. K. Claus, M. Bruns, V. Trouillet, K. Linkert, C. Rodriguez-Emmenegger, A. S. Goldmann, S. Perrier, H. G. Borner and C. Barner-Kowollik, Biomacromolecules, 2013, 14, 4340–4350 CrossRef CAS PubMed.
- L. E. Cheran, D. Vukovich and M. Thompson, Analyst, 2003, 128, 126–129 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb02207j |
| This journal is © The Royal Society of Chemistry 2016 |