
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resolving the α-glycosidic linkage of arginine-rhamnosylated translation elongation factor P triggers generation of the first ArgRha specific antibody†
Xiang
Li‡
a,
Ralph
Krafczyk‡
bc,
Jakub
Macošek
d,
Yu-Lei
Li
ae,
Yan
Zou
a,
Bernd
Simon
d,
Xing
Pan
f,
Qiu-Ye
Wu
a,
Fang
Yan
e,
Shan
Li
f,
Janosch
Hennig
 d,
Kirsten
Jung
bc,
Jürgen
Lassak
*bc and
Hong-Gang
Hu
*a
d,
Kirsten
Jung
bc,
Jürgen
Lassak
*bc and
Hong-Gang
Hu
*a
aDepartment of Organic Chemistry, School of Pharmacy, Second Military Medical University, Shanghai 200433, China. E-mail: huhonggang_fox@msn.com
bDepartment of Biology I, Microbiology, Ludwig Maximilians-Universität München, Munich, Germany
cCenter for Integrated Protein Science Munich, Ludwig-Maximilians-Universität München, Munich, Germany. E-mail: juergen.lassak@lmu.de
dStructural and Computational Biology Unit, EMBL Heidelberg, Heidelberg 69117, Germany
eSchool of Pharmacy, Wei Fang Medical University, Shandong 261053, China
fInstitute of Infection and Immunity, Taihe Hospital, Hubei University of Medicine, Shiyan, Hubei 442000, China
First published on 21st July 2016
Abstract
A previously discovered posttranslational modification strategy – arginine rhamnosylation – is essential for elongation factor P (EF-P) dependent rescue of polyproline stalled ribosomes in clinically relevant species such as Pseudomonas aeruginosa and Neisseria meningitidis. However, almost nothing is known about this new type of N-linked glycosylation. In the present study we used NMR spectroscopy to show for the first time that the α anomer of rhamnose is attached to Arg32 of EF-P, demonstrating that the corresponding glycosyltransferase EarP inverts the sugar of its cognate substrate dTDP-β-L-rhamnose. Based on this finding we describe the synthesis of an α-rhamnosylated arginine containing peptide antigen in order to raise the first anti-rhamnosyl arginine specific antibody (anti-ArgRha). Using ELISA and Western Blot analyses we demonstrated both its high affinity and specificity without any cross-reactivity to other N-glycosylated proteins. Having the anti-ArgRha at hand we were able to visualize endogenously produced rhamnosylated EF-P. Thus, we expect the antibody to be not only important to monitor EF-P rhamnosylation in diverse bacteria but also to identify further rhamnosyl arginine containing proteins. As EF-P rhamnosylation is essential for pathogenicity, our antibody might also be a powerful tool in drug discovery.
Introduction
Glycosylation is one of the most important posttranslational modifications (PTMs) of proteins in biological systems1,2 and is associated with numerous biological processes including viral and bacterial infection, cancer metastasis, inflammatory response, innate and adaptive immunity, as well as many signaling pathways.3,4 For a long time, protein glycosylation was considered to be restricted to eukaryotes. Today it is well accepted that also bacteria including important human pathogens contain a large number of O- and N-linked glycoproteins.5,6 However until 2013 only one case of a sugar being added to arginine was reported.7 At that time, two research groups discovered independently that the type III secretion system effector NleB, of enteropathogenic Escherichia coli (EPEC) acts as arginine-N-acetylglucosamine (ArgGlcNAc) transferase on human death receptor domains, thereby interfering with the host defense.8,9 We elucidated that another type of arginine glycosylation plays an important role in the activation of the specialized translation elongation factor EF-P, which alleviates ribosome stalling at polyproline sequences (Fig. 1).10–13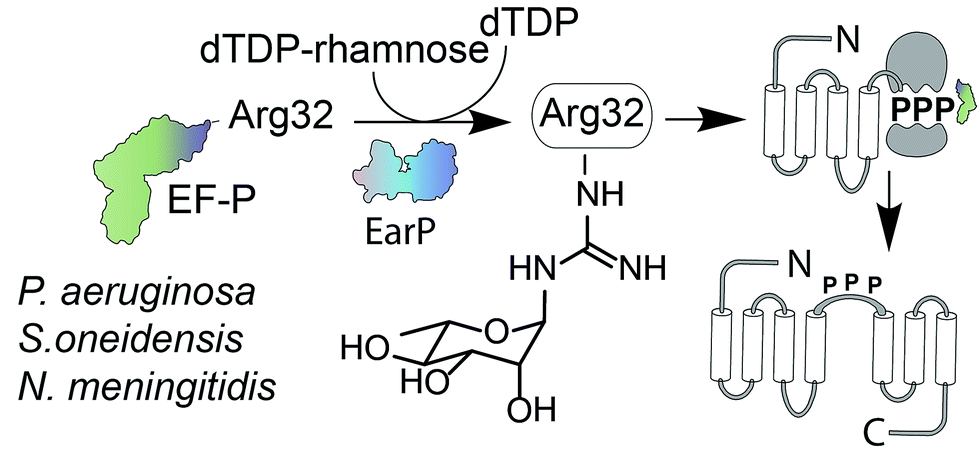 | ||
| Fig. 1 EF-P arginine rhamnosylation and mode of action. Certain bacteria including P. aeruginosa, S. oneidensis, and N. meningitidis encode an EF-P variant with an invariant arginine at position 32. The glycosyltransferase EarP activates EF-P by rhamnosylation of Arg32 using dTDP-β-L-rhamnose as substrate. EF-P and its rhamnose modification stimulate proline–proline peptide bond formation thereby alleviating ribosome stalling at polyproline stretches. EF-P = translation elongation factor P; EarP = EF-P specific arginine rhamnosyl transferase for posttranslational activation. | ||
For effective ribosome rescue certain bacteria, including not only the versatile γ-proteobacterium Shewanella oneidensis MR-1 but also the important human pathogens Pseudomonas aeruginosa and Neisseria meningitidis, post-translationally rhamnosylate a conserved Arg32.14–16 When EF-P is bound to the ribosome the rhamnosylated arginine protrudes towards the peptidyltransferase center thereby contributing to the favorable positioning of the peptidyl-Pro-tRNA and stabilization of the CCA-end of the prolyl-tRNA.14,17,18 Loss of the rhamnose modification abolishes the pathogenicity of P. aeruginosa14 and increases its susceptibility to certain antibiotics.15 Thus inhibition of EF-P rhamnosylation might be a novel strategy to selectively suppress virulence. However, little is known about the corresponding glycosyltransferase EarP or arginine rhamnosylation itself. Here we used NMR spectroscopy and found the rhamnosyl moiety on the protein acceptor EF-P in the α-configuration, unambiguously demonstrating that EarP is an inverting glycosyltransferase. Based on this result, we report the generation of the first high-affinity anti-ArgRha-specific antibody that allowed us to detect rhamnosylated EF-P even from crude cell lysates. With this molecular tool in hand, we will not only be able to improve our understanding of EarP mediated EF-P rhamnosylation, but the antibody might also help to develop new potent targeted antibiotics and to unveil other arginine rhamnosylated proteins.
Results and discussion
EarP mediates an inverting glycosyl transfer reaction
Previously we and others demonstrated that mono-rhamnosylated EF-P-Arg32 in the EarP-arginine phylogenetic subfamily is essential to efficiently alleviate ribosome stalling at polyproline stretches.14–16 However, nothing is known about the anomeric configuration of the attached sugar. Knowledge about steric configuration is important to understand how the modification contributes to the stabilization of the CCA-end of the P-site prolyl-tRNA and to classify EarP either as a retaining or inverting glycosyltransferase.19,20 Notably, the activated sugar substrate is dTDP-β-L-rhamnose. In order to determine the configuration after glycosylation, we employed 13C-edited NOESY-HSQC to assign the sugar resonances (Fig. 2a and b). 1JCH couplings can inform about the configuration of the anomeric carbon. An equatorial position of H1′ (α-anomer) would result in a coupling of around 170 Hz, whereas ≤160 Hz would indicate an axial position (β-anomer).21,22 An undecoupled 13C-HSQC gave rise to a coupling of 167 Hz, clearly indicating an α-configuration on the protein acceptor EF-P (Fig. 2c and d). This was confirmed by the absence of an observable NOE between H1′ and H5′ (Fig. 2b). If H1′ was in the axial position a strong NOE should be visible. The change of configuration at the anomeric center from dTDP-β-L-rhamnose to Arg-α-L-rhamnose during the glycosylation reaction identifies EarP to be an inverting glycosyltransferase.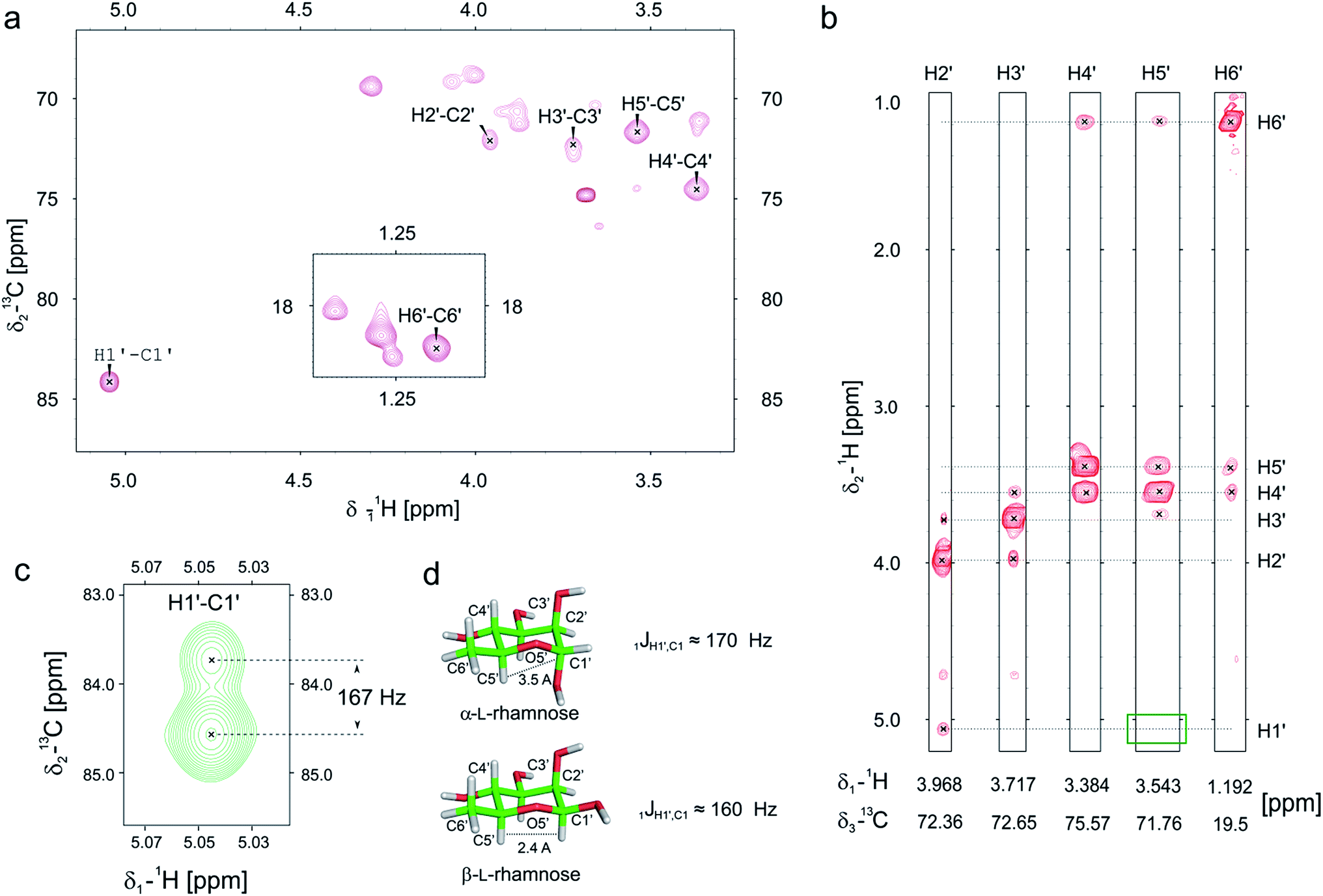 | ||
| Fig. 2 Determination of the EarP rhamnosylation mechanism via NMR. (a) Zoomed in view of the sugar resonance region of the 13C-HSQC of rhamnosylated EF-P. The assignment is based on a 13C-edited NOESY-HSQC (exemplary strips are shown in panel b). Unassigned peaks at around 70 ppm and 18 ppm are the resonances of EF-P's threonine Hβ/Cβ and methyl groups, respectively. (b) Strips of the 13C-edited NOESY-HSQC to illustrate the lack of an observable NOE between H1′ and H5′ (green rectangle), which confirms that the rhamnose adopts an α-configuration, when bound to EF-P. (c) H1′–C1′ resonance of EF-P rhamnose from an undecoupled 13C-HSQC to derive the 1JCH coupling. The resulting coupling of 167 Hz indicates an α-configuration of the sugar.21,22 (d) Stick representations of α-L-, and β-L-rhamnose. | ||
Synthesis of the α-rhamnosylated arginine containing hapten
Having solved the configuration of the rhamnose moiety attached to EF-P-Arg32 we were encouraged to raise specific antibodies against the modification employing an α-rhamnosylated arginine containing peptide, for immunization (Fig. 3a). Such a modification-specific antibody would be a useful tool to investigate EarP mediated rhamnosylation in vivo and in vitro but might also help in the identification of further arginine rhamnosylated proteins from diverse organisms.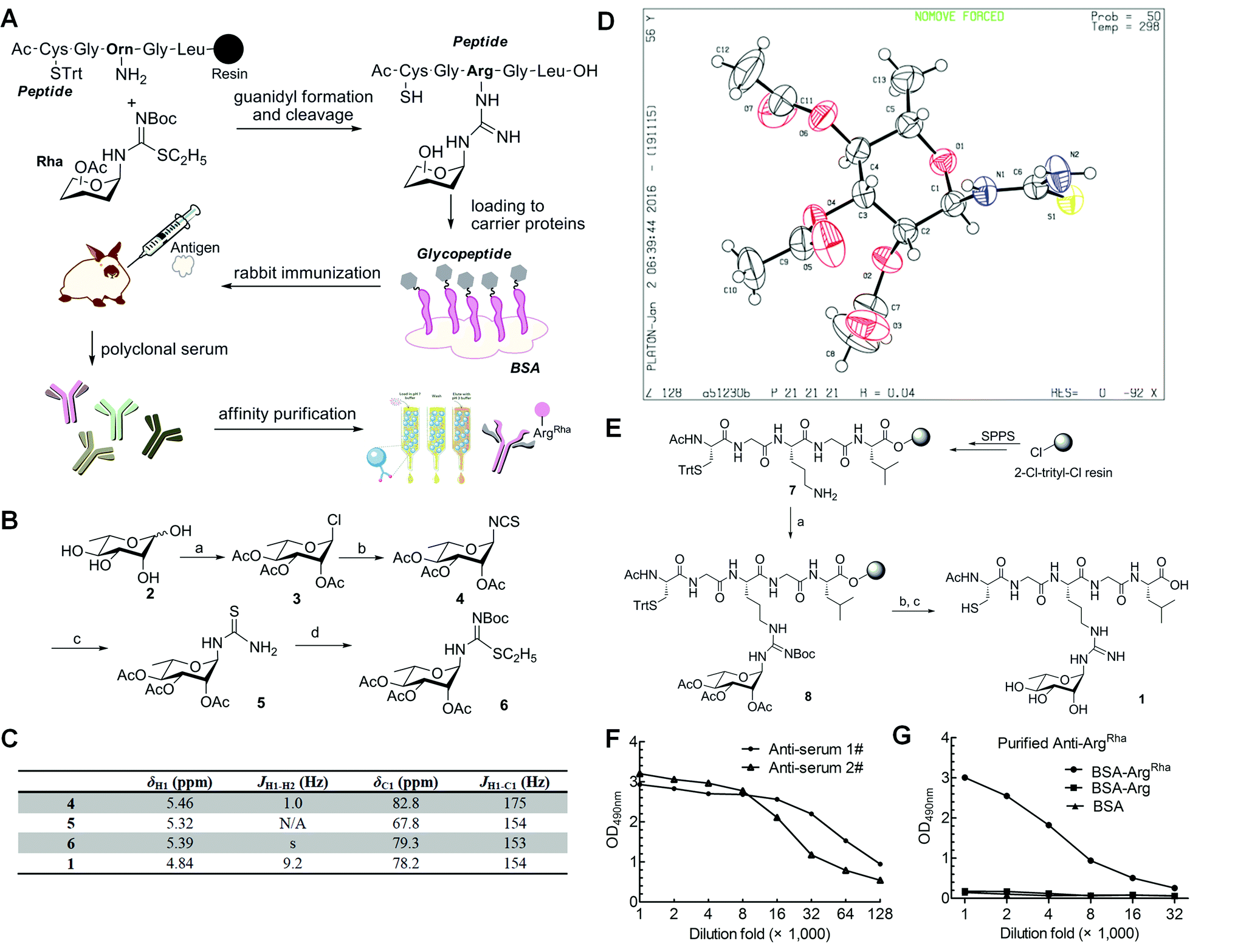 | ||
Fig. 3 Synthesis of mono-ArgRha peptide and antibody generation. (A) Work-flow of antibody generation: in the first step an ArgRha containing glycopeptide was synthesized via guanidyl formation, cleavage and subsequent coupling to bovine serum albumin (BSA). The resulting glycoconjugate was used to immunize rabbits and accordingly to collect crude sera containing polyclonal antibodies against ArgRha. Using a two-step affinity chromatography technique we finally purified a highly sensitive and specific polyclonal anti-ArgRha antibody. Trt = trityl; Boc = tert-butoxycarbonyl. (B) Synthesis of building block 6. Reagents and conditions: (a) acetyl chloride, r.t., 2 days, 85%; (b) KSCN, TBAI, and CH3CN, reflux, 3 h, 70%; (c) NH3, and THF, 1 h, 99%; (d) EtI, and MeOH, reflux, 3 h; then Boc2O, Et3N, and CH2Cl2, 75%. (C) NMR spectroscopic characterization of compounds 4, 5, 6 and 1. (D) Single crystal structure of compound 5. (E) Solid-phase synthesis of mono-ArgRha peptide 1. Reagents and conditions: (a) TEA, DMF, AgNO3, and 6 (3 eq.), r.t.; (b) 5% NH2NH2 in DMF; (c) 5% TIPS in TFA. (F) ELISA analysis of two batches of crude anti-sera. The crude anti-sera immunized by the BSA-glycoconjugate can recognize ArgRha with high affinity. anti-Serum 1# and anti-serum 2# were successively diluted up to 128![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold and subjected to indirect ELISA experiments against the BSA-glycoconjugate. (G) ELISA analysis of purified anti-ArgRha. Purified anti-ArgRha can recognize ArgRha with high specificity. The purified antibody was successively diluted up to 32000 fold and subjected to indirect ELISA experiments against the BSA-glycoconjugate (BSA-ArgRha) and BSA carrying the non-glycosylated peptide (BSA-Arg). 000 fold and subjected to indirect ELISA experiments against the BSA-glycoconjugate. (G) ELISA analysis of purified anti-ArgRha. Purified anti-ArgRha can recognize ArgRha with high specificity. The purified antibody was successively diluted up to 32000 fold and subjected to indirect ELISA experiments against the BSA-glycoconjugate (BSA-ArgRha) and BSA carrying the non-glycosylated peptide (BSA-Arg). | ||
Based on previous work,23,24 we chose a strategy for glycopeptide synthesis that involves direct silver-promoted glycosylation between an S-alkyl-isothiourea and the amine of the amino acid side chain on the solid phase. First, we synthesized the key building block, N-glycosyl-S-alkyl-isothiourea 6, starting from L-rhamnose 2 (Fig. 3b): glycosyl chloride 3 in the desired configuration was obtained using well established procedures (85% yield).25,26 Subsequently, 3 was treated with potassium thiocyanate (KSCN) and tetrabutylammonium hydrogen iodide (TBAI) in anhydrous acetonitrile to get glycosyl isothiocyanate 4 (70% yield).27 Next, glycosyl thiourea 5 was prepared via ammoniation of 4 in tetrahydrofuran (99% yield).28 Finally, a two-step, one-pot procedure converted 5 into 6 in the presence of ethyl iodide and tert-butoxycarbonyl anhydride (75% yield).29 Taken together from 2 to 6 we ended up with an efficiency of about 44%. The configuration of the attached rhamnose in the hapten depends on the stereochemistry of the key intermediate compounds 5 or 6. NMR-HSQC showed that the 1JCH coupling underwent a change from 174 Hz to 154 Hz (Fig. 3c) when compound 4 was converted into 5. Thus we determined the anomeric carbon configuration of compound 5. A single crystal was obtained via slow evaporation of a dichloromethane/n-hexane solution at room temperature (Fig. 3d). With this (N-(2,3,4-tri-O-acetyl-6-deoxy-α-L-manno-pyranos-1-yl)thiourea) in hand, we could show unambiguously that the rhamnose moiety is attached in an α configuration, being consistent with rhamnosylated EF-P. All of the intermediates were fully characterized using 1H-NMR, 13C-NMR, and HR-Q-TOF-MS (Fig. 3c and ESI†).
To synthesize the hapten glycopeptide 1 with the rhamnose moiety in the α-configuration from building block 6, we chose an on-resin glycosylation strategy (Fig. 3e): to obtain the linear peptide we used 9-fluorenylmethyloxycarbonyl (Fmoc) SPPS standard procedures with Fmoc-Orn(Alloc)-OH as the precursor for the ArgRha residue. A 2-chlorotrityl resin acted as the solid support. Subsequent to the peptide assembly, the Alloc group was removed in the presence of tetrakis(triphenylphosphine)palladium (0) to get compound 7 on-resin.30–34 Then the on-resin glycopeptide 8 was synthesized with a silver-promoted solid-phase glycosylation between the free amino group of 7 and the key building block 6. Next, the rhamnose moiety was deacetylated with 5% NH2NH2 in dimethylformamide (DMF).35 Subsequently the resin was treated with 5% triisopropylsilane (TIPS) in trifluoroacetic acid (TFA) to release the glycopeptide 1 which was further purified via preparative reverse-phase HPLC. We calculated from resin loading that the total yield of isolated 1 was 28%, manifesting a good efficiency for the on-resin glycosylation process.36–38 All of the key intermediates were monitored using analytical HPLC and characterized using HR-Q-TOF-MS (Fig. S1†). The final peptide – Cys–Gly–Arg(Rha)–Gly–Leu – was characterized using 1D-NMR, 2D-NMR, and HR-Q-TOF-MS.
Generation and purification of a rhamnosyl arginine specific primary antibody
To raise the high affinity ArgRha specific antibody (anti-ArgRha), the hapten was conjugated to BSA as carrier protein via the free N-terminal sulfhydryl group distal from the arginine rhamnosyl side chain (Fig. 3a). The resulting BSA-glycoconjugate was injected into rabbits to raise polyclonal antibodies targeting the ArgRha moiety.39,40 After the third immunization, the crude anti-sera were collected and their specificity was monitored by employing an enzyme-linked immunosorbent assay (ELISA) analysis with horseradish peroxidase linked anti-rabbit IgG. The antibodies from two batches of crude anti-sera from two immunized rabbits were found to bind robustly and specifically to the BSA-glycoconjugate with high titers, showing strong immune reactivity even after 128000 fold dilution (Fig. 3f and S3†).
To purify anti-ArgRha from the crude rabbit anti sera, in a first step we used a Protein A Sepharose 4 column (Amersham Biosciences). In a second purification step two agarose columns coupled with BSA or BSA carrying the non-glycosylated “naked” pentapeptide (H-CGRGL-OH) were used to exclude cross-reactivity. Taken together, these two steps resulted in a 95% pure anti-ArgRha antibody (Fig. S4 and S5†) showing a significantly improved specificity against the glycoconjugated BSA compared to the crude anti-sera (Fig. 3g and S6†).
anti-ArgRha allows sensitive and specific detection of endogenous EF-PRha
Having the anti-ArgRha at hand, we tested whether we can detect the EF-P rhamnose modification. Therefore we used 0.5 μg of purified EF-P which was modified in vivo (EF-PRha) employing the enzymatic activity of EarP. Unmodified EF-P served as a negative control. As expected, an EF-P specific antibody (anti-EF-P) detected both protein variants with no difference in signal intensity. By contrast anti-ArgRha specifically targeted only the EF-P ArgRha modification and no signal occurred in the lane with unmodified EF-P (Fig. 4a). The amino acid context of the arginine rhamnosylation site in EF-P is Ser30–Gly31–Arg32(Rha)–Asn33–Ala34 and thus significantly differs from the peptide Cys–Gly–Arg(Rha)–Gly–Leu, previously used to raise anti-ArgRha. Thus, we conclude that our antibody recognizes ArgRha irrespective of the adjacent amino acid residues.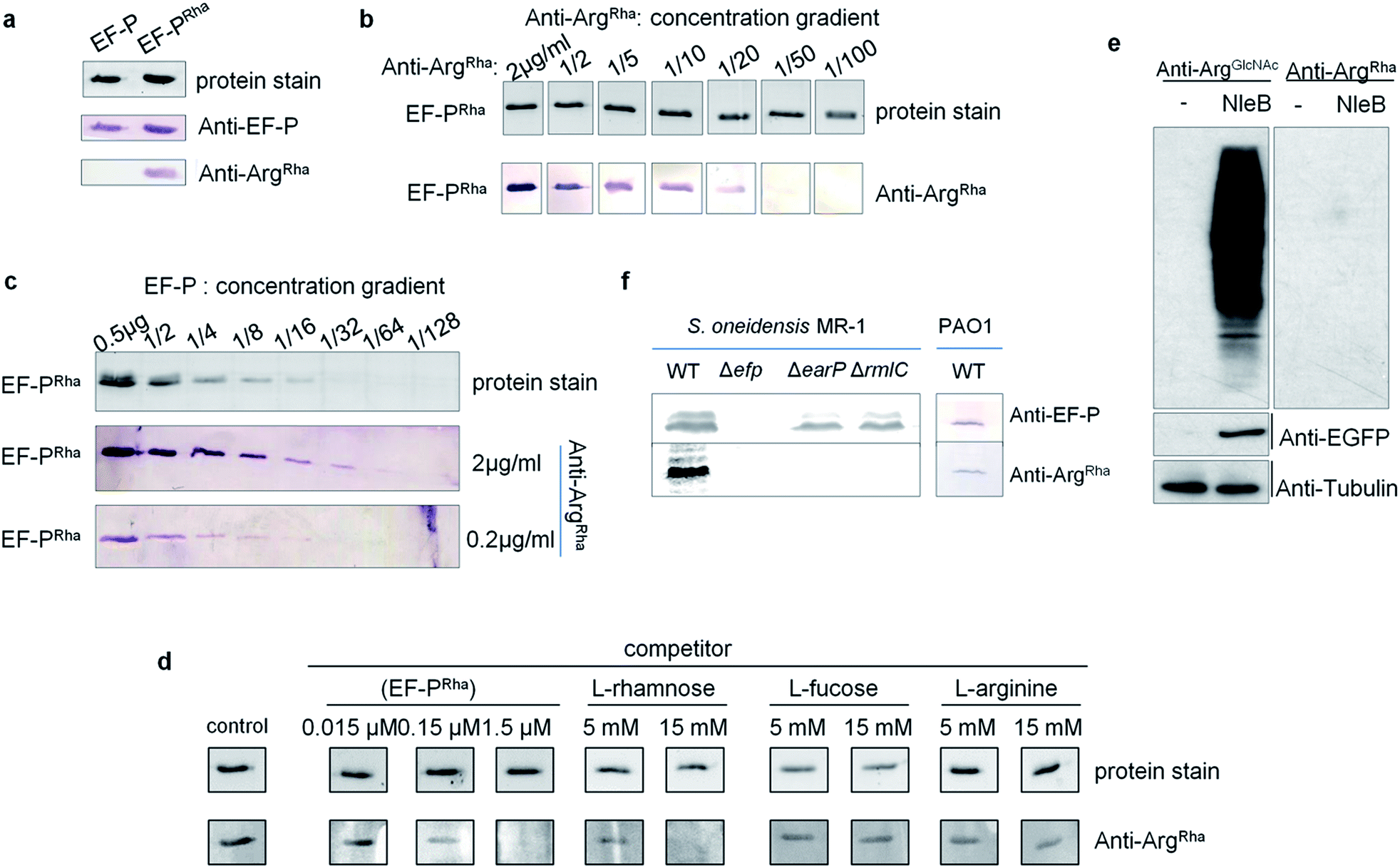 | ||
| Fig. 4 Sensitivity and specificity analysis of anti-ArgRha against EF-PRha. (a) The anti-ArgRha antibody specifically recognizes EF-PRha. Immunodetection of purified EF-P both unmodified (EF-P) and rhamnosylated (EF-PRha) using anti-EF-P and anti-ArgRha. 0.5 μg of purified EF-P was subjected to SDS-PAGE and subsequent Western Blot analysis with 0.2 μg ml−1anti-EF-P or anti-ArgRha respectively. (b) Immunodetection of EF-PRha when anti-ArgRha was successively diluted. (c) Immunodetection of EF-PRha when EF-PRha was successively diluted and anti-ArgRha was used in concentrations of 2 μg ml−1 or 0.2 μg ml−1. (d) Cross-reactivity analysis of anti-ArgRha against L-rhamnose, L-fucose and L-arginine. 0.5 μg of purified EF-PRha were subjected to SDS-PAGE and subsequent Western Blot analysis using 0.2 μg ml−1anti-ArgRha. anti-ArgRha was preincubated with varying concentrations of EF-PRha, L-rhamnose, L-fucose and L-arginine. Buffer only served as a control. (e) anti-ArgRha cannot detect ArgGlcNAc. 293T cells were transfected with mock vector or pCS2–EGFP–NleB plasmids. Western Blot analysis of total cell lysates using either anti-ArgGlcNAc or anti-ArgRha. anti-EGFP and anti-tubulin served as a control. (f) Detection of EF-PRha from S. oneidensis MR-1 lysates of wildtype (WT) and different mutant strains lacking efp (Δefp) the glycosyltransferase EarP (ΔearP) or interfering with dTDP-β-L-rhamnose biosynthesis (ΔrmlC). P. aeruginosa PAO1 WT crude lysates served as an additional in vivo control. Approximately, 108 cells were used per lane. | ||
Next we assessed the detection limits of anti-ArgRha by using varying concentrations of either EF-PRha or anti-ArgRha (Fig. 4b and c). When keeping the EF-PRha concentration constant at 25 μg ml−1 (1.25 μM), the signal intensity progressively decreased starting from 2 μg ml−1anti-ArgRha until no further detection was possible at an antibody concentration of 0.04 μg ml−1. When keeping the anti-ArgRha concentration constant at 2 μg ml−1, 15 ng of EF-PRha were efficiently detected.
To further prove the specificity against ArgRha we performed a Western Blot based competition assay in which our antibody was preincubated with various concentrations of L-rhamnose, L-fucose, or L-arginine. Pre-added EF-PRha served as specific competitor and completely prevented detection of EF-P already at concentrations of 1.5 μM (Fig. 4d). On the contrary even 15 mM of L-arginine or L-fucose could not decrease the signal intensity. At this concentration only L-rhamnose abolished the EF-PRha signal and therefore constitutes a competitor that is around 10000 times less effective than EF-PRha (Fig. 4d and S7†). To examine whether the anti-ArgRha antibody shows cross reactivity towards other types of arginine N-glycosylation, we prepared lysates from 293T cells ectopically expressing NleB. As expected arginine GlcNAcylation could be detected using anti-ArgGlcNAc9,24 but no signal occurred when using anti-ArgRha (Fig. 4e). Taken together, our antibody can be regarded as highly sensitive and specific against arginine rhamnosylation.
We ultimately asked whether we can visualize endogenous arginine rhamnosylated proteins from crude cell lysates. From our sensitivity analysis we calculated that it was possible to detect single ArgRha with about 100 copies per cell when subjecting 108 cells and 2 μg ml−1anti-ArgRha to Western Blot analysis. Rich media exponentially growing E. coli EF-P carry about 10000 copies of EF-P per cell41 and therefore it should be possible to detect the modified protein. As Enterobacteriales modify EF-P with (R)-β-lysine42–44 we used S. oneidensis which naturally employs EarP mediated rhamnosylation. Whereas we could readily identify EF-P in wildtype cells, mutants lacking either efp or earP gave no signal (Fig. 4f). Similarly, we could not detect EF-P rhamnosylation in a strain ΔrmlC that cannot produce the EarP substrate for glycosylation – dTDP-β-L-rhamnose. We used P. aeruginosa PAO1 crude cell lysates to test the activity of the anti-ArgRha antibody in another species and detected a single band (Fig. 4f). The band was verified to be EF-P in a parallel Western Blot, yielding a signal at the same height, by use of a S. oneidensis anti-EF-P antibody. Thus our anti-ArgRha represents a potent tool to detect EF-P rhamnosylation in diverse species.
Conclusion
We recently demonstrated the use of a high affinity anti-N-acetyl glucosaminyl arginine antibody (anti-ArgGlcNAc) to monitor the glycosylation of human death receptor domains mediated by NleB during EPEC infection.9,24 Similarly, anti-ArgRha represents a novel tool to diagnose infections caused by pathogens such as P. aeruginosa14,15 or N. meningitidis.16 Ultimately, our anti-ArgRha might allow us to identify further arginine-rhamnosylated proteins from diverse species. This in turn might help to unveil novel antimicrobial targets and contribute to the task of overcoming the increasing problem of multi resistance. In this regard, it is indispensable to understand the mode of action of arginine dependent glycosyltransferases as they appear to be involved in pathogenicity development. However, our knowledge of N-linked glycosylation is so far mainly restricted to asparagine. The stereospecific outcome of the glycosylation reaction is a major characteristic of its molecular mechanism. By determining the α-anomeric nature of the rhamnosyl moiety on EF-P and with this the inverting mode of glycosyl transfer mediated by EarP, we made the first step to elucidating the catalysis of this novel type of glycosyltransferase. Our finding might also help to further understand how the sugar participates in stabilizing the CCA-end of the P-site prolyl-tRNA and thus contributes to the rescue of polyproline dependent ribosome arrest situations.Author contributions
X. L. performed the organic synthesis, analysis of NMR and single crystal structures of small molecules, generated the anti-ArgRha antibody and wrote the manuscript. R. K. performed the confirmation of antibody specificity, produced proteins for the NMR-analysis of rhamnosylated EF-P and wrote parts of the manuscript. J. M., B. S. and J. H. performed the NMR determination of the anomeric carbon configuration of attached rhamnose and wrote parts of the manuscript. Y.-L. L. assisted in the organic synthesis. Y. Z., Q.-Y. W. and F. Y. assisted in the organic synthesis and antibody generation. X. P. and S. L. assisted in the confirmation of antibody specificity. J. L. performed the confirmation of antibody specificity, contributed to study design and wrote the manuscript. K. J. and H.-G. H. contributed to study design and assisted in modification of the manuscript.Acknowledgements
We thank Instrumental Analysis Center of Second Military Medical University for NMR spectroscopic and mass spectrometric analyses. We are grateful to Ingrid Weitl for excellent technical assistance. J. H. thanks Timmy Fyrner for valuable discussions. This work was supported by the NSFC (No. 21402235), National Major Project of China (2012ZX09J12108-01, 2012ZX09502-001-005) (to X. L., Y. Z., Q.-Y. W. and H.-G. H.), Deutsche Forschungsgemeinschaft Exc114/2, JU270/17-1 (to K. J.), GRK2062 (to K. J. and J. L.). J. H. acknowledges support from the European Molecular Biology Laboratory (EMBL).Notes and references
- W. Yi, P. M. Clark, D. E. Mason, M. C. Keenan, C. Hill, W. A. Goddard III, E. C. Peters, E. M. Driggers and L. C. Hsieh-Wilson, Science, 2012, 337, 975–980 CrossRef CAS PubMed.
- C. Xu, S. Wang, G. Thibault and D. T. Ng, Science, 2013, 340, 978–981 CrossRef CAS PubMed.
- K. R. Mattaini and M. G. Vander Heiden, Science, 2012, 337, 925–926 CrossRef PubMed.
- E. K. Culyba, J. L. Price, S. R. Hanson, A. Dhar, C. H. Wong, M. Gruebele, E. T. Powers and J. W. Kelly, Science, 2011, 331, 571–575 CrossRef CAS PubMed.
- Q. Lu, S. Li and F. Shao, Trends Microbiol., 2015, 23, 630–641 CrossRef CAS PubMed.
- H. Nothaft and C. M. Szymanski, J. Biol. Chem., 2013, 288, 6912–6920 CrossRef CAS PubMed.
- D. G. Singh, J. Lomako, W. M. Lomako, W. J. Whelan, H. E. Meyer, M. Serwe and J. W. Metzger, FEBS Lett., 1995, 376, 61–64 CrossRef CAS PubMed.
- J. S. Pearson, C. Giogha, S. Y. Ong, C. L. Kennedy, M. Kelly, K. S. Robinson, T. W. Lung, A. Mansell, P. Riedmaier, C. V. Oates, A. Zaid, S. Muhlen, V. F. Crepin, O. Marches, C. S. Ang, N. A. Williamson, L. A. O'Reilly, A. Bankovacki, U. Nachbur, G. Infusini, A. I. Webb, J. Silke, A. Strasser, G. Frankel and E. L. Hartland, Nature, 2013, 501, 247–251 CrossRef CAS PubMed.
- S. Li, L. Zhang, Q. Yao, L. Li, N. Dong, J. Rong, W. Gao, X. Ding, L. Sun, X. Chen and F. Shao, Nature, 2013, 501, 242–246 CrossRef CAS PubMed.
- S. Ude, J. Lassak, A. L. Starosta, T. Kraxenberger, D. N. Wilson and K. Jung, Science, 2013, 339, 82–85 CrossRef CAS PubMed.
- L. K. Doerfel, I. Wohlgemuth, C. Kothe, F. Peske, H. Urlaub and M. V. Rodnina, Science, 2013, 339, 85–88 CrossRef CAS PubMed.
- A. L. Starosta, J. Lassak, L. Peil, G. C. Atkinson, K. Virumae, T. Tenson, J. Remme, K. Jung and D. N. Wilson, Nucleic Acids Res., 2014, 42, 10711–10719 CrossRef PubMed.
- L. Peil, A. L. Starosta, J. Lassak, G. C. Atkinson, K. Virumae, M. Spitzer, T. Tenson, K. Jung, J. Remme and D. N. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15265–15270 CrossRef CAS PubMed.
- J. Lassak, E. C. Keilhauer, M. Fürst, K. Wuichet, J. Gödeke, A. L. Starosta, J. Chen, L. Søgaard-Andersen, J. Rohr, D. N. Wilson, S. Häussler, M. Mann and K. Jung, Nat. Chem. Biol., 2015, 11, 266–270 CrossRef CAS PubMed.
- S. J. Hersch, M. Wang, S. B. Zou, K. M. Moon, L. J. Foster, M. Ibba and W. W. Navarre, mBio, 2013, 4, e00180-13 CrossRef PubMed.
- T. Yanagisawa, H. Takahashi, T. Suzuki, A. Masuda, N. Dohmae and S. Yokoyama, PLoS One, 2016, 11, e0147907 Search PubMed.
- G. Blaha, R. E. Stanley and T. A. Steitz, Science, 2009, 325, 966–970 CrossRef CAS PubMed.
- L. K. Doerfel, I. Wohlgemuth, V. Kubyshkin, A. L. Starosta, D. N. Wilson, N. Budisa and M. V. Rodnina, J. Am. Chem. Soc., 2015, 137, 12997–13006 CrossRef CAS PubMed.
- P. M. Coutinho, E. Deleury, G. J. Davies and B. Henrissat, J. Mol. Biol., 2003, 328, 307–317 CrossRef CAS PubMed.
- C. Breton, S. Fournel-Gigleux and M. M. Palcic, Curr. Opin. Struct. Biol., 2012, 22, 540–549 CrossRef CAS PubMed.
- I. Tvaroska and F. R. Taravel, Adv. Carbohydr. Chem. Biochem., 1995, 51, 15–61 CrossRef CAS PubMed.
- W. A. Bubb, Concepts Magn. Reson., 2003, 19A, 1–19 CrossRef CAS.
- X. Li, Y. Li, Y. Chen, Y. Zou, X. Zhuo, Q. Wu, Q. Zhao and H. Hu, RSC Adv., 2015, 5, 94654–94657 RSC.
- M. Pan, S. Li, X. Li, F. Shao, L. Liu and H. Hu, Angew. Chem., Int. Ed., 2014, 53, 14517–14520 CrossRef CAS PubMed.
- A. Singhamahapatra, L. Sahoo, K. J. V. Paul, B. Varghese and D. Loganathan, Tetrahedron Lett., 2013, 54, 6121–6124 CrossRef CAS.
- C. Chang, S. Chang, C. Chao and K. T. Mong, Tetrahedron Lett., 2009, 50, 4536–4540 CrossRef CAS.
- T. K. Lindhorst and C. Kieburg, Synthesis, 1995, 1228–1230 CrossRef CAS.
- A. Kovalová, M. Ledvina, D. Saman, D. Zyka, M. Kubícková, L. Zídek, V. Sklenár, P. Pompach, D. Kavan, J. Bílý, V. Ondřej, K. Zuzana, L. Martina, L. Ljubina, A. Mária, M. Hynek, R. Daniel, H. Kateřina, K. Vladimír and B. Karel, J. Med. Chem., 2010, 53, 4050–4065 CrossRef PubMed.
- D. Ma, C. Xia, J. Jiang, J. Zhang and W. Tang, J. Org. Chem., 2003, 68, 442–451 CrossRef CAS PubMed.
- J. W. Trauger, R. M. Kohli, H. D. Mootz, M. A. Marahiel and C. T. Walsh, Nature, 2000, 407, 215–218 CrossRef CAS PubMed.
- Q. Xiao and D. Pei, J. Med. Chem., 2007, 50, 3132–3137 CrossRef CAS PubMed.
- C. Qin, X. Bu, X. Zhong, N. L. J. Ng and Z. Guo, J. Comb. Chem., 2004, 6, 398–406 CrossRef CAS PubMed.
- C. Qin, X. Bu, X. Wu and Z. Guo, J. Comb. Chem., 2003, 5, 353–355 CrossRef CAS PubMed.
- Y. C. Huang, Y. M. Li, Y. Chen, M. Pan, Y. T. Li, L. Yu, Q. X. Guo and L. Liu, Angew. Chem., Int. Ed., 2013, 52, 4858–4862 CrossRef CAS PubMed.
- C. S. Bennett, S. M. Dean, R. J. Payne, S. Ficht, A. Brik and C. H. Wong, J. Am. Chem. Soc., 2008, 130, 11945–11952 CrossRef CAS PubMed.
- P. Wang, S. Dong, J. H. H. Shieh, E. Peguero, R. Hendrickson, M. A. Moore and S. J. Danishefsky, Science, 2013, 342, 1357–1360 CrossRef CAS PubMed.
- M. N. Amin, J. S. McLellan, W. Huang, J. Orwenyo, D. R. Burton, W. C. Koff, P. D. Kwong and L. X. Wang, Nat. Chem. Biol., 2013, 9, 521–526 CrossRef CAS PubMed.
- J. Hudak, H. Yu and C. Bertozzi, J. Am. Chem. Soc., 2011, 133, 16127–16135 CrossRef CAS PubMed.
- U. Westerlind, H. Schröder, A. Hobel, N. Gaidzik, A. Kaiser, C. Niemeyer, E. Schmitt, H. Waldmann and H. Kunz, Angew. Chem., Int. Ed., 2009, 48, 8263–8267 CrossRef CAS PubMed.
- H. Cai, M. S. Chen, Z. Y. Sun, Y. F. Zhao, H. Kunz and Y. M. Li, Angew. Chem., Int. Ed., 2013, 52, 6106–6110 CrossRef CAS PubMed.
- G. An, B. R. Glick, J. D. Friesen and M. C. Ganoza, Can. J. Biochem., 1980, 58, 1312–1314 CrossRef CAS PubMed.
- T. Yanagisawa, T. Sumida, R. Ishii, C. Takemoto and S. Yokoyama, Nat. Struct. Mol. Biol., 2010, 17, 1136–1143 CAS.
- W. W. Navarre, S. B. Zou, H. Roy, J. L. Xie, A. Savchenko, A. Singer, E. Edvokimova, L. R. Prost, R. Kumar, M. Ibba and F. C. Fang, Mol. Cell, 2010, 39, 209–221 CrossRef CAS PubMed.
- M. Bailly and V. de Crecy-Lagard, Biol. Direct, 2010, 5, 3 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and chemical compound characterization. CCDC 1469830. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02889f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |