
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new perspective in bio-refining: levoglucosenone and cleaner lignin from waste biorefinery hydrolysis lignin by selective conversion of residual saccharides†
M.
De bruyn
a,
J.
Fan
a,
V. L.
Budarin
a,
D. J.
Macquarrie
a,
L. D.
Gomez
b,
R.
Simister
b,
T. J.
Farmer
a,
W. D.
Raverty
c,
S. J.
McQueen-Mason
b and
J. H.
Clark
*a
aThe Green Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK. E-mail: james.clark@york.ac.uk
bThe Centre for Novel Agricultural Products, Department of Biology, University of York, York, YO10 5DD, UK
cDepartment of Chemical Engineering, Monash University, Clayton, Victoria 3800, Australia
First published on 8th July 2016
Abstract
An unexpected opportunity is reported to improve the sustainability of biorefineries whereby 8 wt% levoglucosenone (LGE) can be derived from unconverted saccharides in a lignin-rich biorefinery waste stream in a highly selective fashion (>90%). Additionally, in the process a purer lignin is obtained which can be used for further processing or materials applications. LGE is a valuable and versatile product with a plethora of applications.
Broader contextIf biorefineries are to become economically viable then they will have to become more resilient to the strong price fluctuations of petroleum and be competitive to shale. Also the production cost of bioethanol/biobutanol has to be decreased to below their selling price. Like petro-refineries, bio-refineries need to have a wider product portfolio than just fuels including more valuable chemicals. Here we propose an interesting and new approach to this challenge based on the largely ignored, but potentially very significant residual saccharides in waste biorefinery lignin. Specifically we have shown that these can be converted to the valuable platform molecule levoglucosenone (LGE). Our preferred process involves energy-efficient microwave heating converting 40% of the residual saccharides in the lignin with high selectivity (∼90%), representing an 8 wt% yield based on total lignin. Given the high value of LGE, this could represent a significant income stream. No additives are required and, importantly, the residual lignin is structurally unchanged, making that it can be further depolymerized to aromatics or used in materials applications or as an upgraded fuel. LGE is becoming a valuable intermediate for the production of fuel (5-HMF), the manufacture of new solvents, such as Cyrene™, and other chemicals including monomers e.g. 1,6-hexandiol. |
Current liquid fuel use in road transportation accounts for ∼26% of CO2 emissions to the atmosphere in the US.1 Attempts to substitute and augment fossil liquid transport fuel use with liquids derived from lignocellulosic biomass have been made economically unattractive by overproduction of crude oil, resulting in significant reductions in its market price since 2014.2,3 These economic trends run counter to global initiatives aimed at reducing total greenhouse gas emissions. The resulting low oil price is currently well below the viability threshold of bio-ethanol set at 70–80$ by DuPont4 & 100$ per barrel by the IPCC,5 illustrating how the volatility of oil prices can negatively influence the future of the bioeconomy. Theoretical yields of ethanol from lignocellulosic biomass are plant species dependent and range between 169–370 kg ethanol per dry tonne of biomass.6 The actual yields are considerably lower due to the recalcitrant nature of ligno-cellulosic biomass to enzymatic or chemical hydrolysis, and the difficulty in obtaining concomitant and efficient fermentation of both pentose and hexose sugars.7 At the current market prices for ethanol and 1-butanol, the selling price for the product can easily be absorbed by the cost of the feedstock and its transport, leaving little or no profit margin even before plant operating costs and interest on capital are taken into account. This makes commercial production of ethanol from non-food renewable sources economically unattractive in the absence of government subsidies.8 One way of improving the economic returns from lignocellulosic ethanol production is to valorize the lignin, be it as a polymer, and then potentially part of composites,9 or by converting it to higher value molecules,10 much alike current practice in the petrochemical industry. In the US alone, the projected cellulosic ethanol production of 79 GL by 2022, will generate 62 MT of lignin waste/year.11 Raw lignins after enzymatic hydrolysis and fermentation, however, are known to contain variable amounts of saccharides, reaching up to 30 wt% for softwood.12 We report herein an unexpected opportunity to get value from such waste lignins through conversion of these unconverted saccharides, trapped in the lignin structure after fermentation, to high value levoglucosenone (LGE), while creating in the process a purer lignin. At currently quoted market prices, LGE is ∼70
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times more valuable than ethanol, but even when allowing for a drop in LGE value as market volumes increase, of over 3 orders of magnitude, this discovery has still the potential to change the economics of 2nd generation ethanol production. LGE is also one of the few molecules directly obtainable from lignocellulose with a potential market value that could compete with current non-renewable platform molecules. LGE is typically obtained through pyrolysis of cellulose, in the presence of an acid catalyst. The LGE yields are reported to be strongly dependent on the type of cellulose, its water content, the pretreatment, additives and the catalyst used – a summary is presented in Table S1 (ESI†). Typically ∼10 wt% yield is reported. In the presence of sizeable amounts of ionic liquid, sulfolane or THF, it can reach 20–51 wt%.13–15 However, these methods consume additional resources and yield significant amounts of char. In contrast, the method reported here is simple, consisting only of the microwave (MW) activation of crude waste softwood hydrolysis lignin (CSHL) in air at atmospheric pressure, without additives or specialized handling.
000 times more valuable than ethanol, but even when allowing for a drop in LGE value as market volumes increase, of over 3 orders of magnitude, this discovery has still the potential to change the economics of 2nd generation ethanol production. LGE is also one of the few molecules directly obtainable from lignocellulose with a potential market value that could compete with current non-renewable platform molecules. LGE is typically obtained through pyrolysis of cellulose, in the presence of an acid catalyst. The LGE yields are reported to be strongly dependent on the type of cellulose, its water content, the pretreatment, additives and the catalyst used – a summary is presented in Table S1 (ESI†). Typically ∼10 wt% yield is reported. In the presence of sizeable amounts of ionic liquid, sulfolane or THF, it can reach 20–51 wt%.13–15 However, these methods consume additional resources and yield significant amounts of char. In contrast, the method reported here is simple, consisting only of the microwave (MW) activation of crude waste softwood hydrolysis lignin (CSHL) in air at atmospheric pressure, without additives or specialized handling.
Conventional pyrolysis is a potentially simple route to convert the saccharides in CSHL into valuable compounds. To probe this, dried CSHL, obtained after pilot scale simultaneous saccharification and fermentation, was pyrolized conventionally in a combined thermogravimetric analyzer and infrared spectrometer (TG-IR) under a N2 flow. Fig. 1A shows the FT-IR spectrum of the volatile fraction generated at 310 °C. While the occurrence of 5-hydroxymethylfurfural (5-HMF), formic acid, formaldehyde, CO2 and H2O is well known, the formation of LGE (Fig. 1A and B), in up to 6 wt% yield (lignin based), was unexpected (Fig. S1–S4, ESI†). In view of LGE's tendency to form 5-HMF at high temperature,16 it was assumed that a lower operating temperature could increase the LGE yield. We have previously shown that MW activation can enable reaction temperatures up to 100 °C lower than those needed for conventional pyrolysis processes.17 MW treatment of CSHL to 180 °C, typically requiring less than 5 min total reaction time, produced an increase in the LGE yield from 6 to 8 wt%. More specifically 9 wt% (459 mg) physically recovered oil with 90% LGE purity was obtained, representing 40 wt% based on residual saccharides (see also ESI†). The purity is calculated from GC-MS (Fig. 1D) and can also be inferred from the 13C NMR spectra of the bio-oil (Fig. 1C). The MW route was also applied to softwood pulp/sawdust impregnated with 0.5 wt% H2SO4 but no LGE could be obtained. Some potential explanations for this observation are listed in ESI.†
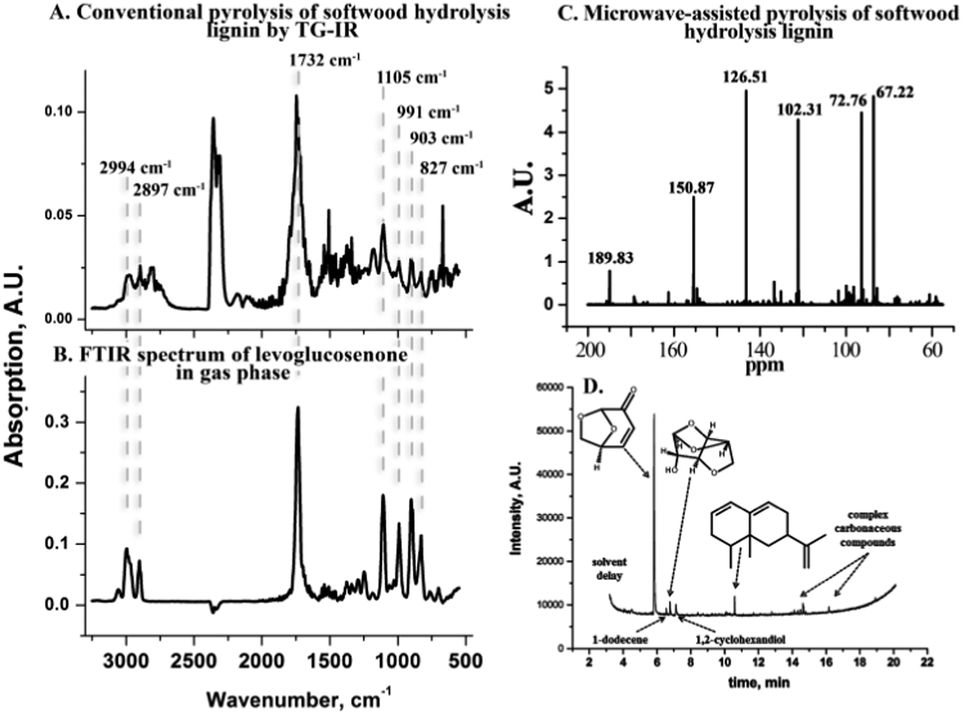 | ||
| Fig. 1 (A) FT-IR spectrum of the volatiles from conventional pyrolysis of crude waste softwood hydrolysis lignin (CSHL) at 310 °C. (B) Gas phase FT-IR spectrum of LGE. (C) 13C NMR spectrum of the liquid extract from MW-assisted pyrolysis of CSHL with labelled LGE peaks. (D) GC-MS of the liquid extract from the MW-assisted pyrolysis of CSHL. | ||
LGE has multiple applications, such as a precursor of 1,6-hexanediol and 1,2,6-hexanetriol. These are key intermediates in the industrial synthesis of 1,6-hexanediamine, caprolactone and caprolactam, which are used for the manufacturing of polyester, polyamide and polyurethanes, representing multimillion tonnes operations.18,19 LGE can also be isomerized into 5-HMF, a valuable precursor for both pharmaceuticals and fuels.20 Recently, dihydro-LGE has been reported as a safe replacement for the reprotoxic solvents NMP and DMF21 and its production is now at pilot plant scale (1 T per week) reflecting strong industrial interest (see ESI†). The chiral nature of LGE also lends itself to the synthesis of natural products (Fig. 2).22
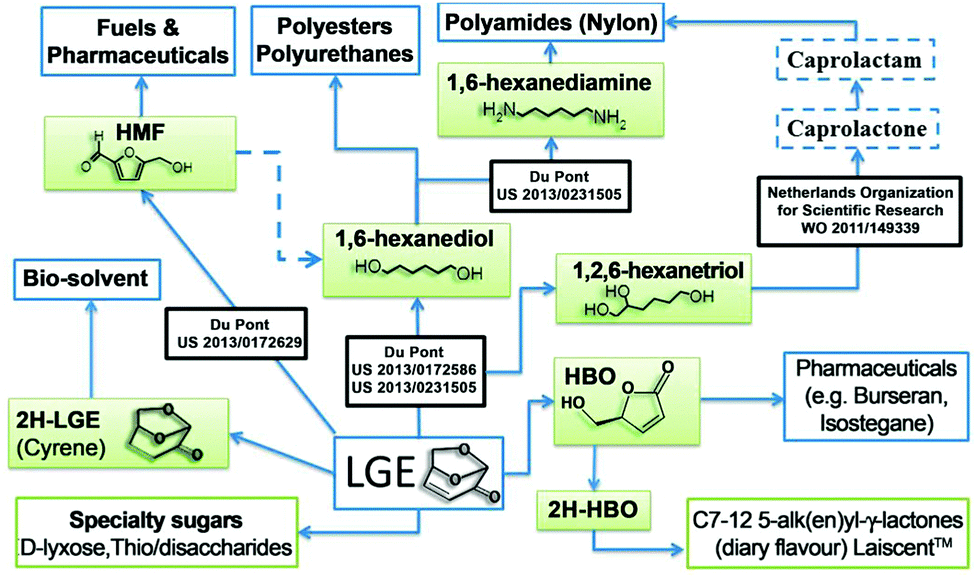 | ||
| Fig. 2 Prospective applications of LGE. | ||
The amount and composition of the unconverted saccharides in CSHL were determined using a standard procedure23 (Fig. 3A). The water-soluble saccharide fraction present in CSHL was determined at 11.2 wt%, consisting mainly of glucose (45 wt%) and cellobiose (54 wt%). The insoluble fraction was hydrolysed with 2 M trifluoroacetic acid, solubilizing non-crystalline polysaccharides and giving 0.64 wt% as mostly glucose (60.9 wt%). Subsequently, the remaining insolubles were subjected to H2SO4 hydrolysis (consecutive 72 wt% and 3.2 wt%) to determine the crystalline components (8.9 wt%), alongside glucose (59 wt%) and mannose (28 wt%) (see ESI†). These analyses show the presence of a range of saccharides from which LGE can be derived, notably cellulose, cellobiose and glucose.15,24,25 As the analyzed CSHL is the product of a high temperature pretreatment of biomass, no clear distinction can be made between the saccharides coming from hemicellulose or cellulose, explaining the high mannose content from the H2SO4 hydrolysis step. In addition, GC-MS of acetone extracts of the CSHL (Fig. S5, ESI†) did not reveal any LGE that may have resulted from the high temperature acid pretreatment step in the biorefinery. XPS analysis of CSHL revealed a C/O ratio similar to those reported in the literature for lignin.26 Residual acidity and its type was probed by FT-IR using pyridine titration (Fig. 3C–i). The difference spectrum (Fig. 3C–ii) reveals the pyridinium salt peaks. The absorption bands at 1645 and 1552 cm−1 confirm the presence of Brønsted acidity, while those at 1615 and 1450 cm−1 reveal some Lewis acidity.27 We attribute the Lewis acidity to an accumulation of (plant) metal species during the various steps of the biorefinery process (see ESI†). Nine consecutive washes of CSHL leaches ∼0.34 wt% H2SO4 (Fig. S6, ESI†). XPS (Fig. 3B) and ICP-MS allow for the determination of 0.78 and 0.63 wt% S which is markedly more than the 0.11 wt% S from the acid titration. This discrepancy may be due to Na2SO4, from the neutralization step in the bioethanol process, or potentially some sulfonated lignin. MW pyrolysis of acid depleted CSHL shows the formation of both LGE and levoglucosan (LGA) (Fig. S7, ESI†), which is an important piece of evidence that LGA is in this case (see ESI†) the precursor to LGE.28
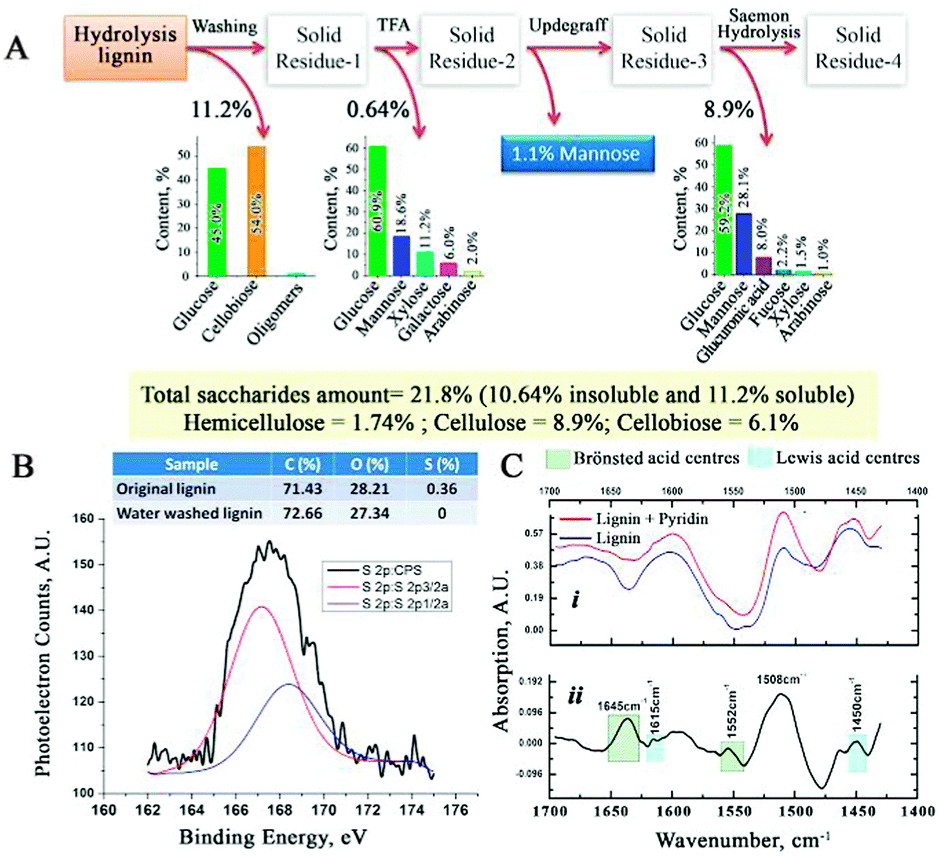 | ||
| Fig. 3 (A) Saccharide composition of CSHL (B) XPS of CSHL (C) FT-IR of CSHL before and after pyridine titration. | ||
We then investigated the physical structure of the CSHL. SEM of original and fully washed CSHL revealed the presence of an irregular macro- and mesoporosity (Fig. 4A and B). This likely reflects the zones where cellulose fibers have been removed by the action of acid and enzymes. N2 adsorption of washed and original CSHL gave type IV isotherms confirming the presence of mesopores (Fig. 4C). Using the BJH method a broader pore distribution, centering on 11 nm radius, was calculated (Fig. 4D). Interestingly literature data show that typical cellulase enzymes are unable to diffuse into pores less than 20–30 nm radius, implying that the enzymes could only reach and convert the hydrolyzed saccharides present in the larger pores.29 Original CSHL shows a markedly lower pore volume (Fig. 4C), which can be explained by their partial blockage with mono- and oligosaccharides. We propose that LGE is formed from dispersed saccharides in the lignin matrix in the presence of residual H2SO4. These saccharides may be adsorbed or bonded covalently to the lignin matrix, potentially even residing within enzyme-inaccessible mesopores (<30 nm). Once LGE is formed during the heating step (MW/thermal) it is rapidly removed to the macroporous network where fast diffusion out of the material takes place, avoiding further reaction. Crucial to the enhanced LGE yield/selectivity under MW operation is the cooler gas phase as MWs cannot directly heat a gaseous medium. Additionally, the presence of elevated amounts of lignin and the full or partial elimination of hemicelluloses can favor LGE formation.30 CSHL fulfills these conditions as the saccharide content has been significantly decreased in the biorefinery step, eliminating nearly all hemicellulose, meanwhile increasing the lignin content. Importantly it is known that LGE can be removed by distillation from biomass residues. CSHL before and after MW treatment was analyzed by FT-IR (Fig. 4E and F). It can be seen that this reduces markedly the saccharide C–OH vibrations at ∼1000–1100 and ∼3200–3500 cm−1, while leaving distinct lignin vibrations at 1698 (normalized), 1587 and 1507 cm−1 unaltered.31 This suggests little alteration of the CSHL before and after MW treatment, which is consistent with the higher temperature experienced by the CSHL in the biorefinery. Also, after MW processing the C–OH vibrations are still clearly visible, and display a cellulosic signature, suggesting that the unconverted saccharides are largely intact and not converted to coke. Fig. S8 and S9 (ESI†) show the DSC traces of CSHL respectively in its unwashed original form and after washing. Original unwashed CSHL displays a small thermal event in the broader 180–220 °C zone which disappears after washing. This may be linked to water soluble saccharides rather than a lignin component. Besides the 458.6 mg recovered LGE, the MW process also produces 4.1 g purer lignin. A mass balance and CHN analysis have been included in ESI.†
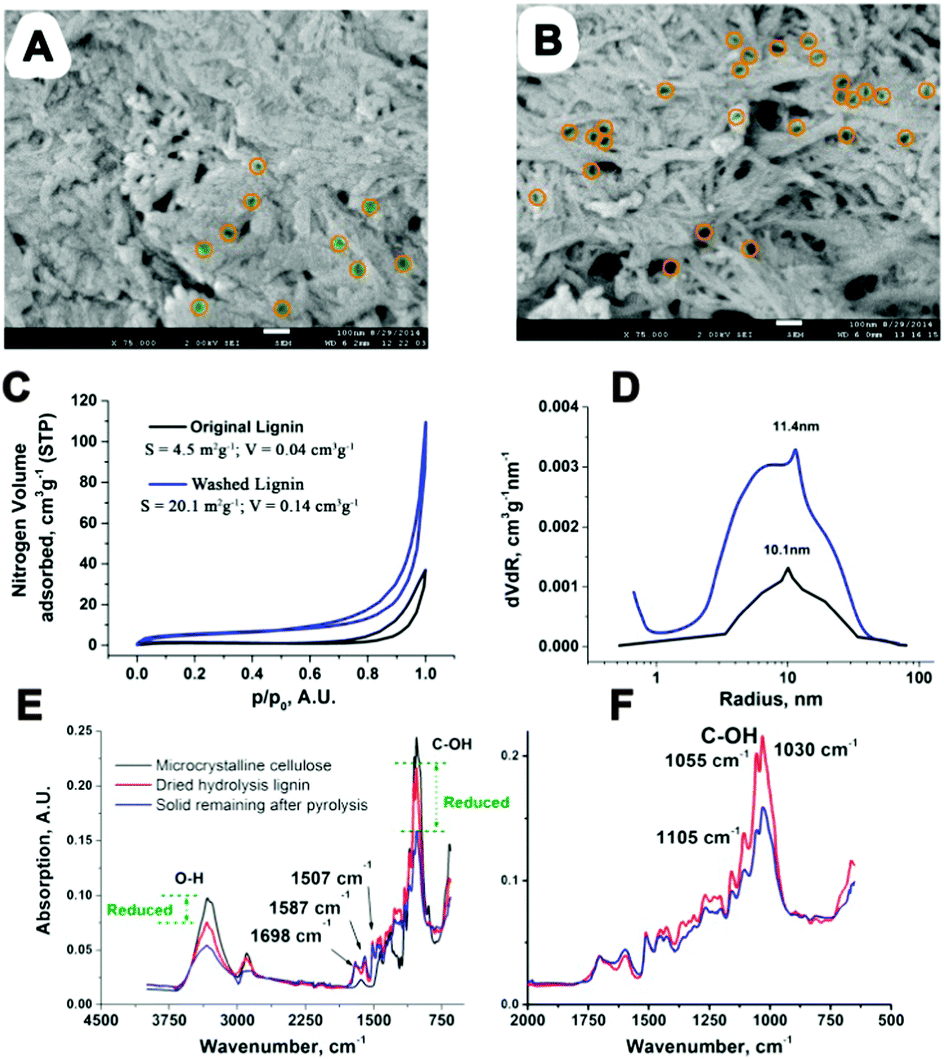 | ||
| Fig. 4 (A) SEM of original CSHL x75000 with yellow encircled mesopores (B) SEM of washed CSHL x75000 with indicated mesopores (C) N2 adsorption isotherms of washed/original CSHL. (D) Pore distribution derived from N2 adsorption data for washed/original CSHL. (E and F) FT-IR of original CSHL (dried), the solid remaining after MW heating of CSHL and subsequent extraction of LGE (dried solid) and microcrystalline cellulose. | ||
Conclusions
We report the production of LGE from CSHL, a waste product of bioethanol production, while also producing a purer lignin. In contrast to existing methods the LGE formation does not require the need of pure or anhydrous cellulose, the use of additives and/or the use of organic solvents. Furthermore, the process takes place at atmospheric pressure, removing the need for high pressure reactors. LGE is likely derived from zones in the lignin matrix where the saccharides were previously inaccessible for enzymes. Yields are increased to 8 wt% LGE (lignin-based) by the lower MW operating temperature. This equals 40 wt% yield based on the residual saccharides. It is also shown that the MW operation has a very limited impact on the lignin structure so it is still available for further use/processing. Many methods are currently being developed to utilize lignin,9,32 and combined with LGE production, they could significantly improve the economic viability of biorefineries. It is envisaged that this technology will be easily scalable using existing commercial scale MW tunnel heating technology of the type used to dry food.Acknowledgements
We thank: SP Processum AB for financial support; Domsjö Fabriker & SEKAB for the CSHL, EPSRC for research grant no. EP/K014773/1 and the European Union's FP7 Research and Innovation funding programme for grant no 251132 (SUNLIBB).Notes and references
- Sources of Greenhouse Gas Emissions, http://www3.epa.gov/climatechange/ghgemissions/sources/transportation.
- Crude Oil Price History Chart, http://www.macrotrends.net/1369/crude-oil-price-history-chart.
- K. Wong, OUTLOOK '16: The year the world drowned in oil, 2015 Search PubMed.
- E. Crooks, Biofuel needs $70 oil to compete, says DuPont, 2015 Search PubMed.
- H. Chum, A. Faaij,J. Moreira, G. Berndes, P. Dhamija, H. Dong, B. Gabrielle, A. Goss Eng, W. Lucht, M. Mapako, O. Masera Cerutti, T. McIntyre, T. Minowa and K. Pingoud, Special Report on Renewable Energy Sources and Climate Change Mitigation, 2011, p. 284 Search PubMed.
- U. D. O. Energy, Alternative Fuels Data Center: theoretical ethanol yields of selected feedstocks (table), http://www.afdc.energy.gov/fuels/ethanol_feedstocks.html.
- P. E. Marriott, L. D. Gomez and S. J. McQueen-Mason, New Phytol., 2016, 209, 1366–1381, DOI:10.1111/nph.13684.
- J. Clemente, Why Biofuels Can't Replace Oil, http://www.forbes.com/sites/judeclemente/2015/06/17/why-biofuels-cant-replace-oil.
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef CAS.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709–720 CrossRef CAS PubMed.
- M. L. Rabinovich, Cellul. Chem. Technol., 2010, 44, 173–186 CAS.
- S. Kudo, Z. Zhou, K. Norinaga and J.-I. Hayashi, Green Chem., 2011, 13, 3306–3311 RSC.
- H. Kawamoto, S. Saito, W. Hatanaka and S. Saka, J. Wood Sci., 2007, 53, 127–133 CrossRef CAS.
- F. Cao, T. Schwartz, D. McClelland, S. Krishna, J. Dumesic and G. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 CAS.
- Y. C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, J. Phys. Chem. C, 2009, 113, 20097–20107 CAS.
- V. L. Budarin, P. S. Shuttleworth, J. R. Dodson, A. J. Hunt, B. Lanigan, R. Marriott, K. J. Milkowski, A. J. Wilson, S. W. Breeden, J. J. Fan, E. H. K. Sin and J. H. Clark, Energy Environ. Sci., 2011, 4, 471–479 CAS.
- A. M. Allgeier, N. Desilva, E. Korovessi, C. Menning, J. C. Ritter and S. K. Sengupta, US Pat., 20130172629, 2013 Search PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melian-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- J. C. Ritter and C. S. Stauffer, US Pat., 20130172580, 2013 Search PubMed.
- J. Sherwood, M. De Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- A. M. Sarotti, M. M. Zanardi, R. A. Spanevello and A. G. Suarez, Curr. Org. Synth., 2012, 9, 439–459 CrossRef CAS.
- L. Jones, J. L. Milne, D. Ashford and S. J. McQueen-Mason, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11783–11788 CrossRef CAS PubMed.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, J. Anal. Appl. Pyrolysis, 2009, 86, 323–330 CrossRef CAS.
- Q. Lu, Y. Zhang, C.-Q. Dong, Y.-P. Yang and H.-Z. Yu, J. Anal. Appl. Pyrolysis, 2014, 110, 34–43 CrossRef CAS.
- G. Henriksson, J. Li, L. Zhang and M. E. Lindström, in Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals, ed. M. Crocker, Royal Society of Chemistry, Cambridge, 2010, ch. 9, pp. 222–262 Search PubMed.
- R. Luque, J. M. Campelo, D. Luna, J. M. Marinas and A. A. Romero, Microporous Mesoporous Mater., 2005, 84, 11–20 CrossRef CAS.
- Y.-C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, J. Phys. Chem. C, 2009, 113, 20097–20107 CAS.
- P. Bubner, J. Dohr, H. Plank, C. Mayrhofer and B. Nidetzky, J. Biol. Chem., 2012, 287, 2759–2765 CrossRef CAS PubMed.
- J. Zandersons, A. Zhurinsh, G. Dobele, V. Jurkjane, J. Rizhikovs, B. Spince and A. Pazhe, J. Anal. Appl. Pyrolysis, 2013, 103, 222–226 CrossRef CAS.
- I. Santoni, E. Callone, A. Sandak, J. Sandak and S. Dire, Carbohydr. Polym., 2015, 117, 710–721 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee01352j |
| This journal is © The Royal Society of Chemistry 2016 |