
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coinage metal complexes with bridging hybrid phosphine–NHC ligands: synthesis of di- and tetra-nuclear complexes†‡
Thomas
Simler
a,
Pierre
Braunstein
*a and
Andreas A.
Danopoulos
*ab
aLaboratoire de Chimie de Coordination, Institut de Chimie (UMR 7177 CNRS), Université de Strasbourg, 4 rue Blaise Pascal, 67081 Strasbourg Cedex, France
bInstitute for Advanced Study (USIAS), Université de Strasbourg, 4 rue Blaise Pascal, 67081 Strasbourg Cedex, France. E-mail: braunstein@unistra.fr; danopoulos@unistra.fr
First published on 11th February 2016
Abstract
A series of P–NHC-type hybrid ligands containing both PR2 and N-heterocyclic carbene (NHC) donors on meta-bis-substituted phenylene backbones, LCy, LtBu and LPh (R = Cy, tBu, Ph, respectively), was accessed through a modular synthesis from a common precursor, and their coordination chemistry with coinage metals was explored and compared. Metallation of LPh·n(HBr) (n = 1, 2) with Ag2O gave the pseudo-cubane [Ag4Br4(LPh)2], isostructural to [Ag4Br4(LR)2] (R = Cy, tBu) (T. Simler, P. Braunstein and A. A. Danopoulos, Angew. Chem., Int. Ed., 2015, 54, 13691), whereas metallation of LR·HBF4 (R = Ph, tBu) led to the dinuclear complexes [Ag2(LR)2](BF4)2 which, in the solid state, feature heteroleptic Ag centres and a ‘head-to-tail’ (HT) arrangement of the bridging ligands. In solution, interconversion with the homoleptic ‘head-to-head’ (HH) isomers is facilitated by ligand fluxionality. ‘Head-to-tail’ [Cu2Br2(LR)2] (R = Cy, tBu) dinuclear complexes were obtained from LR·HBr and [Cu5(Mes)5], Mes = 2,4,6-trimethylphenyl, which also feature bridging ligands and heteroleptic Cu centres. Although the various ligands LR led to structurally analogous complexes for R = Cy, tBu and Ph, the rates of dynamic processes occurring in solution are dependent on R, with faster rates for R = Ph. Transmetallation of both NHC and P donor groups from [Ag4Br4(LtBu)2] to AuI by reaction with [AuCl(THT)] (THT = tetrahydrothiophene) led to LtBu transfer and to the dinuclear complex [Au2Cl2LtBu] with one LtBu ligand bridging the two Au centres. Except for the silver pseudo-cubanes, all other complexes do not exhibit metallophilic interactions.
Introduction
Phosphine and NHC donors are often compared because they readily coordinate to metal centres and display bonding analogies and tuneable stereo-electronic properties.1 However, despite the fact that both are considered as strong σ-donors, emerging evidence reveals subtly different σ-donating and π-accepting properties, diversifying across the periodic table.2 This can lead to transition metal complexes with beneficial catalytic properties, e.g. finely controlled lability and metal electronic tuning, stability of the catalytically active species etc. The complementary roles of both types of donors participating in the same metal coordination sphere may enhance synergism,3 although counter examples have been described.4 The beneficial synergism may be enhanced if the hetero donors are part of a hybrid ligand. This background justifies synthetic efforts towards the design of new phosphine-functionalised NHC (P–NHC) complexes,5 with reported high activities in C–C coupling reactions (PdII, IrI),6 amination of aryl chlorides (PdII)7 and transfer hydrogenation of ketones (RuII).8Among the P–NHC-type ligands, bidentate hybrid ligands with direct P–N bond,9 flexible alkyl,6a–c,f,8,10 or more rigid and tuneable aryl spacer between the donors, 1a and 1b, respectively, have attracted most attention (Fig. 1);6d,7,11 in particular, we and others have been interested in the meta-bis-substituted phenylene framework 1c–1d as potential precursor to non-symmetrical PCCNHC ‘pincer’ complexes.12 Relevant PCNHCP pincer and P2(CNHC)2 macrocyclic ligands 26f,9c,13 and 3,5a respectively, have been described.
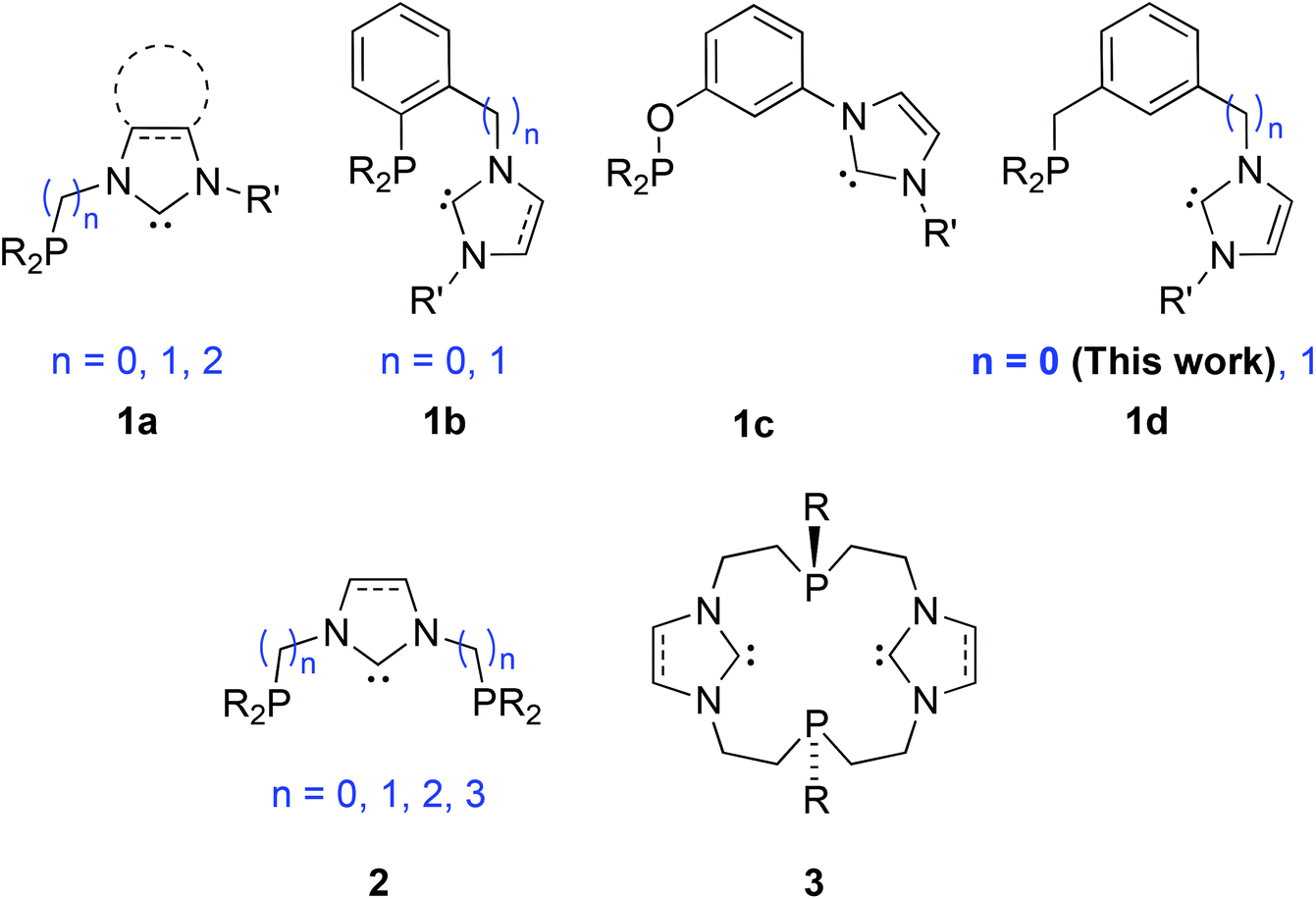 | ||
| Fig. 1 Bidentate P-NHC-type (1a–1d), tridentate PCNHCP (2) and macrocyclic P2(CNHC)2 (3) hybrid ligands reported. | ||
The coordination chemistry of P–NHC-type ligands has mainly been focussed on late transition metals; the few structurally characterized examples14 incorporating AgI,9a,c,10g,11a,12a,c,e,15 or CuI are depicted in Fig. 2.9c,10f,12e,13b,15c This relative scarcity is surprising, considering the interest for air stable group 11 NHC complexes. Silver P–NHC complexes are usually obtained by the reaction of the corresponding imidazolium salts with Ag2O,16 or by initial formation of the free carbene ligand followed by coordination to AgI.9c,15c In addition to their structural diversity, they have proved to be efficient NHC transfer reagents to metals,16 such as RuII,15a,17 RhI,11a,18 PdII,6f,10g,13a and AuI;9a,15c,19 but in rare cases the transmetallation did not proceed neatly.10g,12c
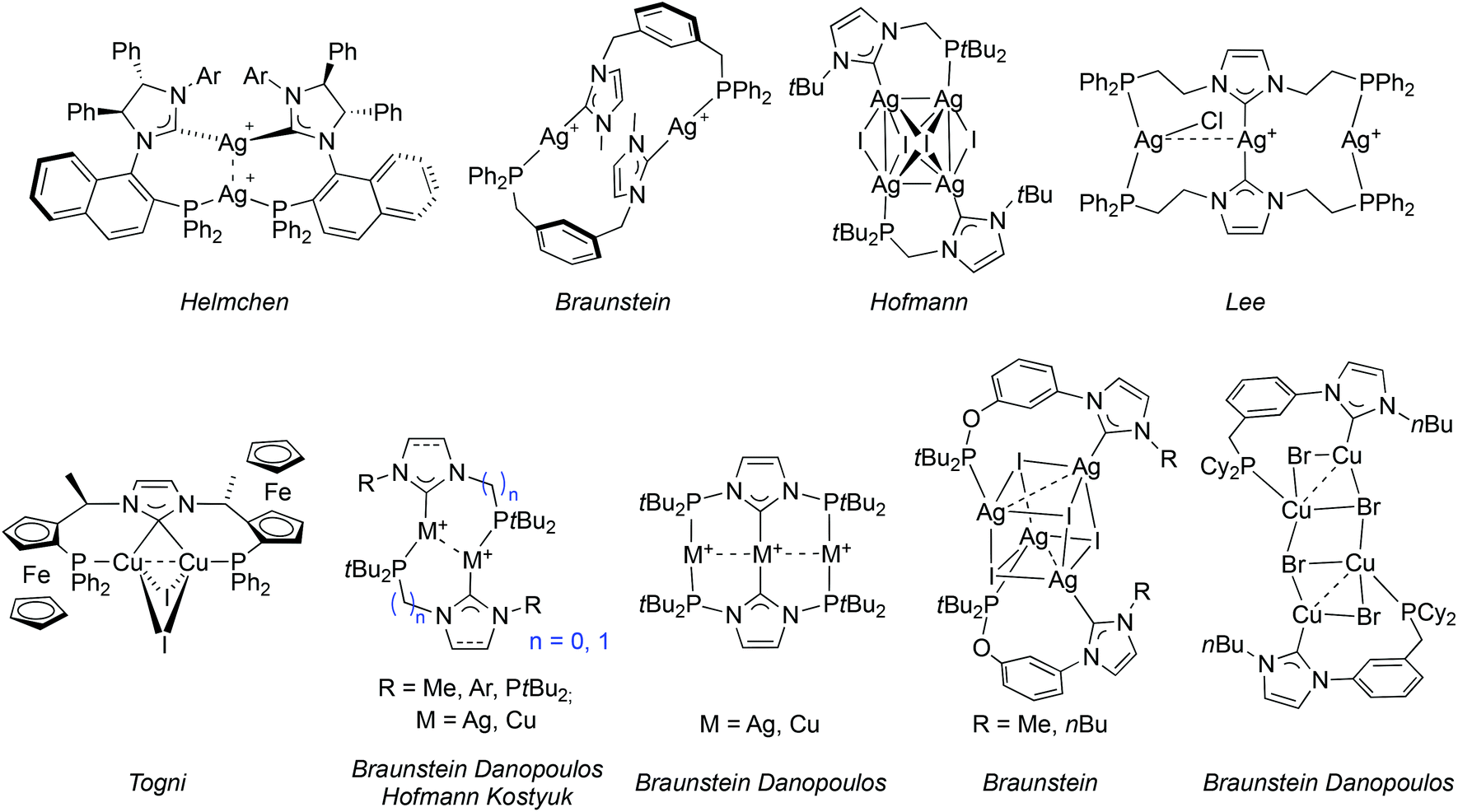 | ||
| Fig. 2 Structurally characterised P–NHC-type silver(I) and copper(I) complexes reported in the literature. | ||
Interestingly, P–NHC-type copper(I) complexes are accessible by transmetallation from the corresponding AgI complexes,9a,12e,15c and by other methodologies e.g. the coordination of the pre-formed free carbene to a labile CuI precursor,9c,10f,13b,15c or the reaction of the imidazolium salt with precursors featuring a coordinated base (e.g. copper(I) acetate,13b mesitylcopper(I) [Cu5(Mes)5]12e and [CuN(SiMe3)2]).9c
Lastly, P–NHC gold(I) complexes are scarce (Fig. 3) but arouse increasing interest due to their attractive photophysical properties and the occurrence of metallophilic interactions in their structures.9a,c,e,15c,19
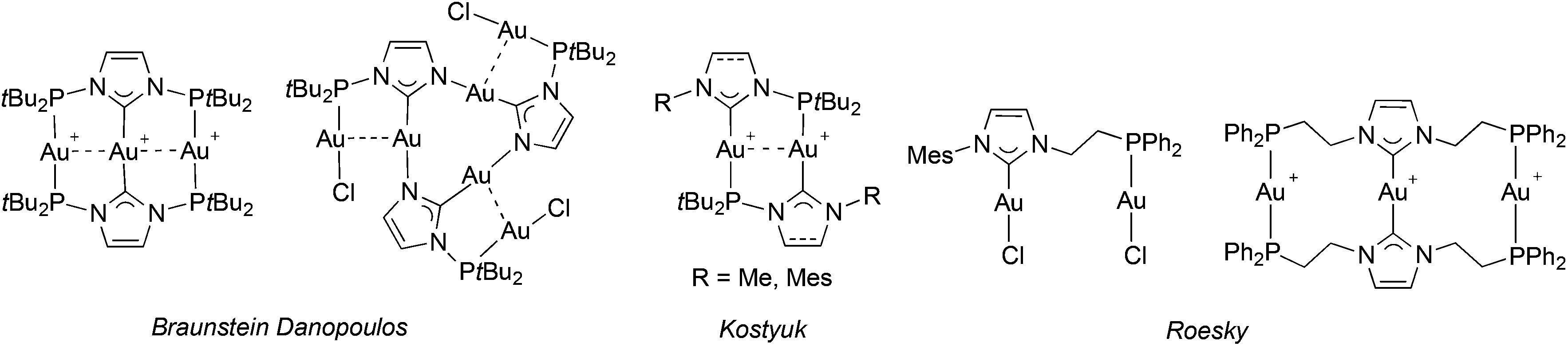 | ||
| Fig. 3 Gold(I) complexes with P–NHC-type ligands reported in the literature (all since 2013). | ||
Extending our previous work on P-based NHC hybrid ligands,12a,c,e herein we report an efficient and modular access to the RP–NHC-type (RP = PCy2, PtBu2 or PPh2) ligands (see 1d in Fig. 1) and their coinage metal complexes.
Results and discussion
Ligand synthesis
A synthetic strategy for the synthesis of phosphine imidazolium precursors employing silane (SiHMeCl2 or SiHCl3) reduction20 of readily available phosphoryl-imidazolium salts has ample literature precedence,6b,10a,15b,21 including attempted preparation of precursors of similar topology to those described below.12c This methodology requires the use of excess silane reductants and forcing conditions, usually leading to moderate yields. Therefore, an alternative, wider scope synthetic strategy was developed, that is easily adaptable to different phosphine substituents (Scheme 1).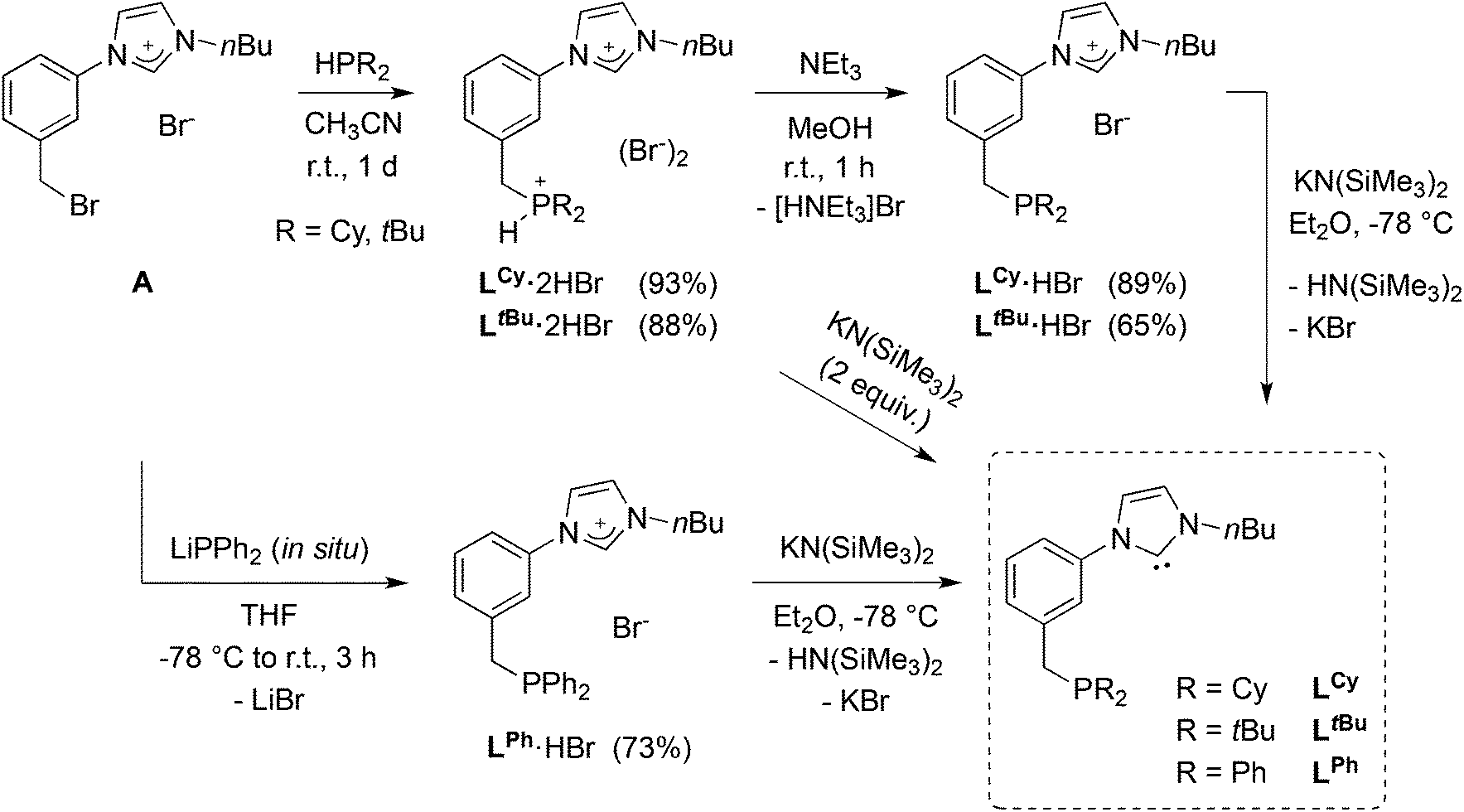 | ||
| Scheme 1 Introduction of phosphine moieties to obtain hybrid P-NHC-type ligands. The synthesis of LCy·2HBr and LtBu·2HBr has been reported in a previous communication.12e | ||
Starting from the imidazolium–bromobenzyl derivative A, the air-stable phosphonium–imidazolium salts LCy·2HBr and LtBu·2HBr were obtained by quaternisation of dicyclohexyl- and di-tert-butylphosphine in acetonitrile,12e and converted to the corresponding phosphine-imidazolium salts LCy·HBr and LtBu·HBr by treatment with NEt3. Successful single deprotonation was confirmed in the 1H-NMR spectra by the disappearance of the deshielded signal due to the acidic P–H proton (1JP–H ≈ 480–490 Hz). Singlets at δ 5.8 and 32.0 ppm for LCy·HBr and LtBu·HBr, respectively, were observed in the 31P{1H}-NMR spectra. Due to the relative air-sensitivity of the trialkyl-phosphine products, borane-protection of the phosphine in LCy·HBr was carried out and yielded LCy·HBr·BH3 as an air-stable crystalline solid, the structure of which is shown in Fig. 4 (left).
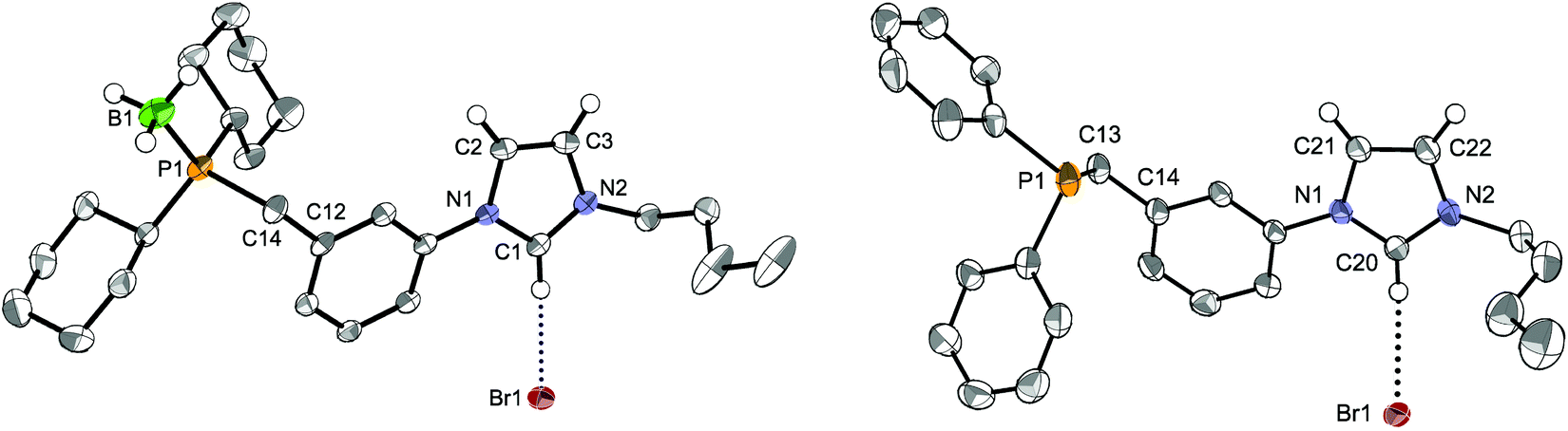 | ||
| Fig. 4 The molecular structures of the borane-protected LCy·HBr·BH3 (left) and of LPh·HBr (right) with thermal ellipsoids at 40% probability. For clarity, only one disordered nBu chain in LCy·HBr·BH3 is displayed and H atoms have been omitted, except for the imidazolium and borane moieties. Selected bond distances (Å) and angles [°] for LCy·HBr·BH3: B1–P1 1.914(3), C1–N1 1.342(3), C1–N2 1.318(3), C1H⋯Br1 3.511(2); N1–C1–N2 108.6(2), P1–C14–C12 117.9(2); for LPh·HBr: C20–N1 1.334(3), C20–N2 1.321(3), C20H⋯Br1 3.598(3); N1–C20–N2 108.7(2), P1–C13–C14 111.5(2). | ||
When an analogous synthetic route was applied to LPh·HBr, it failed in the step of the direct quaternisation of diphenylphosphine by A owing to the lower nucleophilicity of the former. To circumvent the problem, lithium diphenylphosphide (LiPPh2), generated in situ, was reacted with A at low temperature (Scheme 1). Formation of LPh·HBr was confirmed by a phosphorus resonance at δ −8.5 ppm. In the different LR precursors, the imidazolium backbone protons usually gave rise in the 1H-NMR spectra to apparent triplets (overlapping dd, 3JHH ≈ 4JHH ≈ 1.6–1.8 Hz), and the NCHN signal was observed in the range δ 11.17–11.46 ppm.
In the structure of the moderately air-stable LPh·HBr (Fig. 4, right) the imidazolium and central aryl ring planes form an angle of 13.4° (vs. 22.6° for LCy·HBr·BH3). Other bond distances and angles are unremarkable. H-bonding interactions in the solid state were evidenced by a close contact between the NCHN proton and the bromide anion, in addition to the high directionality of the C–H⋯Br− interaction.22 Anion metathesis of LPh·HBr and LtBu·HBr with excess NaBF4 resulted in the corresponding LPh·HBF4 and LtBu·HBF4 salts (see Experimental section). In their 1H-NMR spectra, the signal of the NCHN proton appeared shifted upfield (δ 9.05 and 9.18 ppm, respectively),23 consistent with weaker hydrogen bonding compared to the bromide salts.
Formation of the free carbenes
The free carbenes LCy, LtBu and LPh were obtained by the double deprotonation of the corresponding phosphonium–imidazolium LR·2HBr or the single deprotonation of phosphine–imidazolium LR·HBr salts with stoichiometric amounts of KN(SiMe3)2 (Scheme 1). The free carbenes were obtained in high yields (79–90%) as very air sensitive, pentane soluble, dark green oils that turned red on standing for ca. 30 min at room temperature. The reason for such colour change is still unclear but probably linked to thermal and/or photochemical instability, however, the products of decomposition were not identified. Despite the difficulties associated with the long-term storage and handling of the isolated LCy, LtBu and LPh, unequivocal spectroscopic evidence for their identity and purity was obtained. Deprotonation and carbene formation was evidenced by the disappearance of the imidazolium signal in the 1H-NMR spectra of the oils and the observation of the NCN carbene resonance at δ 215.9–216.2 ppm.24 Due to the difficult handling of LR, the synthesis of the coinage metal complexes described below is based on reactions with the imidazolium salt precursors LR·n(HBr) (n = 1, 2).Synthesis and structure of silver complexes
The availability of LR·HBr opened the way for a comparative study of the coordination chemistry of LR as a function of R. Treatment of LPh·HBr with 1 mole equiv. of Ag2O in acetonitrile, in the presence of 4 Å molecular sieves, afforded [Ag4Br4(LPh)2] in low yields (<50%) after recrystallization from CH2Cl2/Et2O (Scheme 2, route (a)). Upon formation of the silver complex, the disappearance of the signal due to the acidic imidazolium proton and the downfield shift of the broad singlet at δ 3.2 ppm in the 1H-NMR and 31P-NMR spectrum, respectively, confirmed NHC formation and coordination of the P atom. The absence of P–Ag couplings (107Ag 51.8% and 109Ag 48.2%, both I = 1/2) can be rationalised by a dynamic behaviour involving rapid P–Ag bond breaking/formation on the 31P-NMR timescale.12e In the 13C-NMR spectrum, the coordinated CNHC was detected as a broad singlet (Δν1/2 = 12 Hz) at δ 186.5 ppm, in the typical range for NHC–Ag complexes.25 The absence of 13C–107/109Ag coupling has been reported in related NHC–AgX clusters,12a,c,23,26 and points towards dynamic behaviour in solution27 and a high lability of the NHC–Ag bond.16,28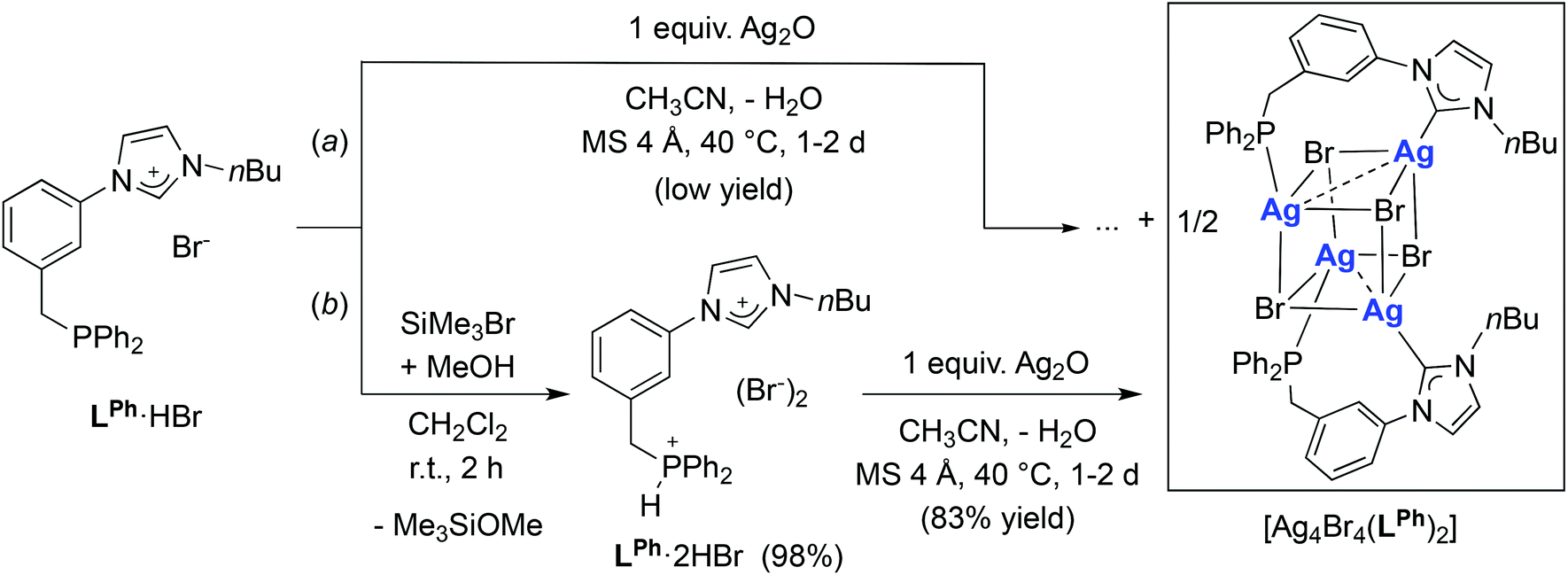 | ||
| Scheme 2 Synthesis of the silver complex [Ag4Br4(LPh)2] (yields based on LPh). | ||
The structure of [Ag4Br4(LPh)2] (Fig. 5) corresponds to a distorted Ag4Br4 cubane cluster with alternating vertices of the cube being occupied by Ag and Br atoms. The two bridging LPh-κP,κCNHC ligands each span the Ag⋯Ag diagonal of two parallel Ag2Br2 faces of the cube, forming 9-membered dimetallocycles, as observed with a closely related phosphinite–NHC ligand12a,c and in the structures of [Ag4Br4(LR)2] (R = Cy, tBu) recently reported.12e All bromides are capping three Ag centres. The Ag⋯Ag separations (3.300(1) Å and 3.400(1) Å) are shorter than the sum of the van der Waals radii for Ag (3.44 Å),29 implying weak d10–d10 interactions.30 Related [Ag4(halide)4L2] cubane structures have been described with L = phosphine ligands,31 and recently obtained with bidentate ligands incorporating NHC donors (bis-NHC23,26,32 or P–NHC-type10g,12a,c,e ligands). Containing non-symmetrical ligands, the observed molecular structure is chiral due to the lack of an improper axis of rotation (see Fig. 6); however, the two enantiomers are present in the asymmetric unit (related by the inversion centre of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ).
).
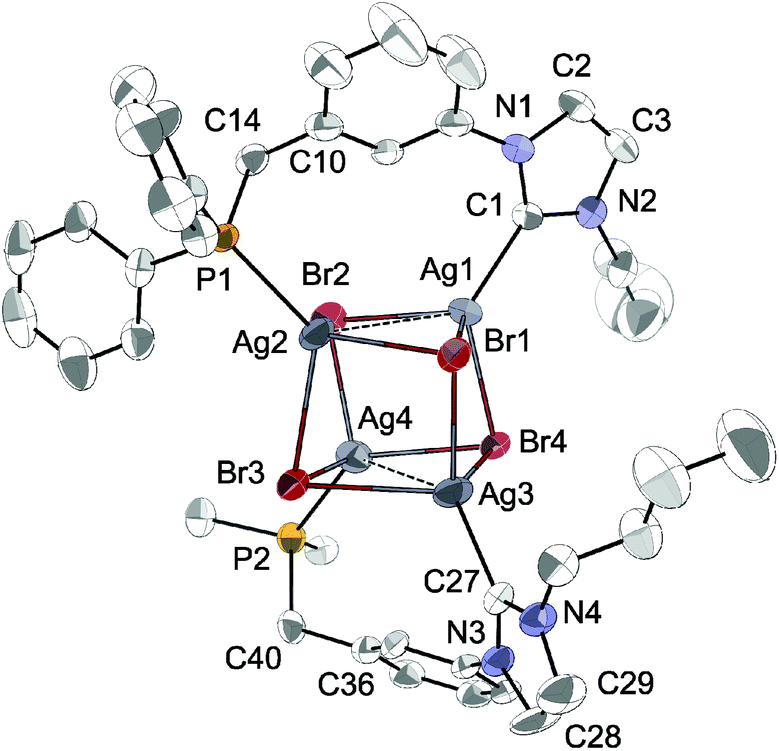 | ||
| Fig. 5 The molecular structure of [Ag4Br4(LPh)2] with thermal ellipsoids at 40% probability. For clarity, H atoms are omitted and only the ipso carbons of the phenyl substituents in the lower ligand are shown. Selected metrical data are given in Table 1. | ||
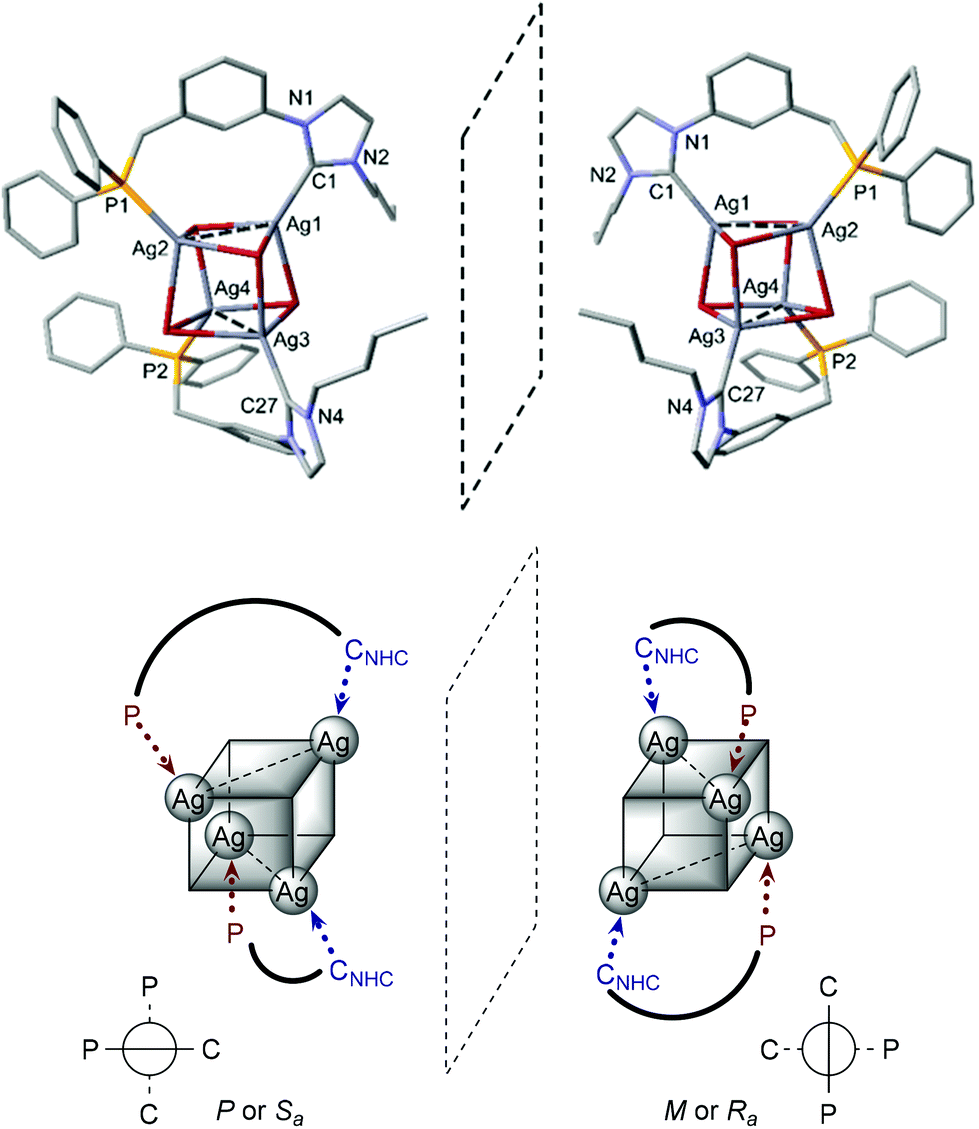 | ||
| Fig. 6 Schematic representation of the two enantiomers of [Ag4Br4(LPh)2] in the crystallographic unit cell. | ||
Comparison of [Ag4Br4(LPh)2] with the previously reported structures of [Ag4Br4(LR)2] (R = Cy, tBu)12e reveals that the substituents on the phosphorus have little influence on the adopted motif or the metrical data. For example, with LPh and LCy, the Ag–CNHC and Ag–P bond distances are comparable, while Ag–P is marginally longer in [Ag4Br4(LtBu)2] (difference <0.040 Å). A more notable difference is in the Ag⋯Ag separation in each bridged face of the pseudocubane (mean Ag⋯Ag ca. 3.350 Å for [Ag4Br4(LPh)2], 3.188 Å for [Ag4Br4(LCy)2] and 3.089 Å for [Ag4Br4(LtBu)2]), leading to complexes with increased distortion from the idealised cubane geometry, which may be ascribed to intramolecular repulsions of the bulkier P-substituents.33 Comparative metrical data for the different silver complexes are provided in Table 1.
| [Ag4Br4(LtBu)2]a | [Ag4Br4(LCy)2]a | [Ag4Br4(LPh)2] | [Ag2(LPh)2](BF4)2·2CH2Cl2 | [Ag2(LtBu)2](BF4)2·CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
|
|---|---|---|---|---|---|
| a Data taken from ref. 12e. b There are two dinuclear complexes exhibiting similar metrical data in the asymmetric unit, the second set of values refers to the other molecule. | |||||
| Ag1⋯Ag2 | 3.101(1) | 3.188(1) | 3.400(1) | 5.361(1) | 5.508(1)/5.762(1) |
| Ag3⋯Ag4 | 3.076(1) | 3.188(1) | 3.300(1) | ||
| Ag1⋯Ag3 | 3.821(1) | 3.721(1) | 3.761(1) | ||
| Ag1⋯Ag4 | 3.712(1) | 3.687(1) | 3.562 (1) | ||
| Ag1–Br1 | 2.688(1) | 2.880(1) | 2.949(1) | ||
| Ag1–Br2 | 3.006(1) | 2.812(1) | 2.748(1) | ||
| Ag1–Br4 | 2.895(1) | 2.708(1) | 2.721(1) | ||
| Ag2–P1 | 2.422(1) | 2.402(1) | 2.407(1) | Ag1–P2 2.386(2) | Ag1–P2 2.376(2)/2.357(2) |
| Ag4–P2 | 2.425(1) | 2.391(1) | 2.388(1) | Ag2–P1 2.376(2) | Ag2–P1 2.375(2)/2.395(2) |
| Ag1–C1 | 2.148(4) | 2.135(3) | 2.147(5) | Ag1–C1 2.113(6) | Ag1–C1 2.106(6)/2.094(6) |
| Ag3–C27 | 2.123(4) | 2.144(4) | 2.127(5) | Ag2–C27 2.108(6) | Ag2–C23 2.109(7)/2.113(7) |
| N1–C1–N2 | 103.7(4) | 103.9(3) | 103.7(5) | 104.6(1) | 104.1(5)/104.7(5) |
| N3–C27–N4 | 103.2(3) | 103.3(3) | 103.1(5) | 105.1(5) | 103.1(6)/102.5(6) |
In view of the similarity between [Ag4Br4(LPh)2] and [Ag4Br4(LR)2] (R = Cy, tBu), the latter having been obtained from the corresponding phosphonium–imidazolium salts, we reasoned that better yields of [Ag4Br4(LPh)2] should also be attainable by the reaction of LPh·2HBr with one mole equiv. Ag2O. Indeed, the reaction of LPh·2HBr with Ag2O afforded the expected cubane complex in very good yields (>80%). It is worth noticing that the method of choice for the preparation of LPh·2HBr consisted of protonation of LPh·HBr by dry HBr, generated in situ by methanolysis of an exactly stoichiometric amount of SiMe3Br in dichloromethane under oxygen-free, controlled conditions (Scheme 2, route (b)). We also noted that the reaction of LPh·HBr with 0.5 mole equiv. Ag2O in acetonitrile resulted in the formation of another silver complex featuring 1H NMR resonances distinct from [Ag4Br4(LPh)2], the structure of which remains elusive to date.
The crucial role of halides in the formation of the cubane structures described above raised the question of the possible reaction outcome under halide-free conditions. The reaction of LPh·HBF4 with 0.5 mole equiv. of Ag2O in acetonitrile led to the complex [Ag2(LPh)2](BF4)2 (Scheme 3). Examination of its 1H and 31P{1H} NMR spectra revealed an equilibrium involving two isomers in solution. Notably, dissolution in CD3CN gave rise, in the 31P{1H} NMR spectrum, to two sets of two doublets (total 8 lines) observed at δ 21.3 (two doublets, 1JP–107Ag ≈ 500 Hz, 1JP–109Ag ≈ 580 Hz) and 11.2 ppm (two doublets, 1JP–107Ag ≈ 475 Hz, 1JP–109Ag ≈ 550 Hz) in a 1:1.1 ratio, respectively. Evaporation of the solvent and re-dissolution in CD2Cl2 led to a similar set of peaks but in a ca. 4:1 ratio, respectively. The reversibility of this procedure confirmed the solvent-dependency of the equilibrium. Due to limited solubility in CD3CN, the 13C{1H}-NMR spectrum was recorded in CD2Cl2, where only the signals for the major isomer were clearly visible. In order to gain more insight into the structures of these two isomers, crystallisations from either CH2Cl2 or CH3CN solutions were attempted. Products corresponding to [Ag2(LPh)2](BF4)2·(solvent)x were obtained from both solvents, which crystallized in different space groups as ‘head-to-tail’ (HT) (heteroleptic) isomers with respect to the mutual arrangement of the ligands. However, the molecular structure of the products (Fig. 7, left and Fig. S1 in ESI‡) revealed the same atom connectivity and very similar metrical data, indicating that only one and the same isomer crystallised (with a possible shift of the equilibrium between ‘head-to-head’ (HH) (homoleptic) and HT isomers upon crystallisation).
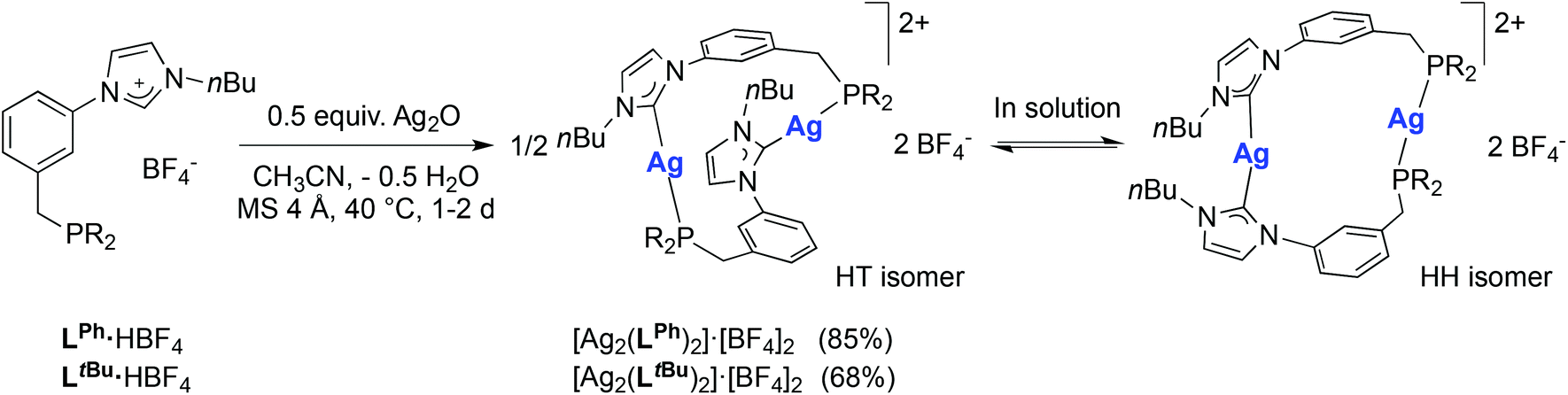 | ||
| Scheme 3 Synthesis of the silver complexes [Ag2(LR)2](BF4)2 (R = Ph, tBu) and ‘head-to-tail’ (HT)/‘head-to-head’ (HH) isomerisation in solution (see text). Yields are based on LR. | ||
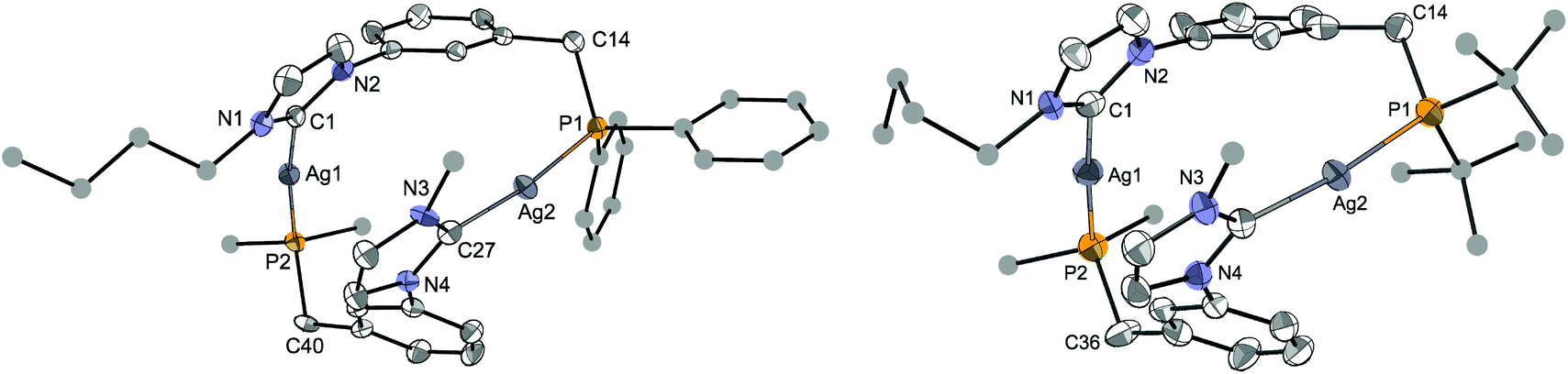 | ||
| Fig. 7 Structure of the dication in [Ag2(LPh)2](BF4)2·2CH2Cl2 (left) and of one of the two dications in [Ag2(LtBu)2](BF4)2·CH2Cl2 (right), with thermal ellipsoids at 40% probability. Anions, H atoms and crystallisation solvent are omitted for clarity. C atoms for the Ph, tBu and nBu groups are depicted as spheres of arbitrary radii (only one C atom is displayed for these groups in the lower ligands). Selected metrical data are given in Table 1. The structure of the [Ag2(LPh)2](BF4)2 complex, obtained by crystallisation from CH3CN, is presented in the ESI.‡ | ||
In the structure of [Ag2(LPh)2](BF4)2·2CH2Cl2 (Fig. 7, left), the two LPh ligands bridge two Ag metal centres (Ag1⋯Ag2 5.361(1) Å) in a ‘head-to-tail’ arrangement. The CNHC–Ag–P angles slightly deviate from linearity (C1–Ag1–P2 172.2(2)° and C27–Ag2–P1 172.7(2)°) and the two NHC rings are not parallel, their mean planes forming an angle of 12.8°. Such an arrangement has already been observed in other P–NHC-type silver complexes;10g,12c,15b the linear coordination geometry is also encountered in bis-NHC silver complexes with non-coordinating anions.23,34 The Ag–CNHC bond distances follow trends observed for related complexes,25a being slightly longer in the NHC silver–halide clusters (mean ca. 2.137 Å)14 than in the complexes with non-coordinating anions (mean ca. 2.111 Å).14
In order to gain insight into the solution behaviour of [Ag2(LPh)2](BF4)2, the corresponding [Ag2(LtBu)2](BF4)2 was similarly prepared (Scheme 3). In this case too, 1H- and 31P{1H}-NMR analysis in CD2Cl2 revealed the presence of two isomers, in a 1:2 ratio, the nature of which could be determined by perusal of the 13C{1H}-NMR spectrum. Spectra of sufficient quality were obtained by acquisition with a cryogenically cooled probe head. A complex pattern (10 lines in total) in the region δ 180–177 ppm, corresponding to the CNHC–Ag signals was successfully simulated, revealing two different CNHC–Ag environments associated with the different isomers (Fig. 8): the two doublets centred at δ 178.8 ppm (1JC–107Ag = 183 Hz, 1JC–109Ag = 212 Hz) were attributed to an isomer with homoleptic AgI centres and symmetrical NHC–Ag–NHC coordination (HH isomer), while two doublets of doublets at δ 178.5 ppm (1JC–107Ag = 190 Hz, 1JC–109Ag = 219 Hz, 2JP–Ag–C = 62 Hz) were assigned to the second and major isomer, with heteroleptic NHC–Ag–P connectivity (HT isomer). Further indication of the nature of the former isomer was obtained from the observation in 13C-NMR of ‘virtual’ triplets of the XnAA′X′n (X = X′ = C, A = A′ = P) spin system involving the carbon atoms directly bound to phosphorus, resulting from a strong 2JPAgP coupling between trans-coordinated P donors.35 Interestingly, for all [Ag2(LR)2](BF4)2 (R = Ph, tBu) complexes, the 1H-NMR signals for the NHC backbone protons were detected as apparent triplets, likely due to 4JHAg and 3JHH coupling constants falling in the same range.36
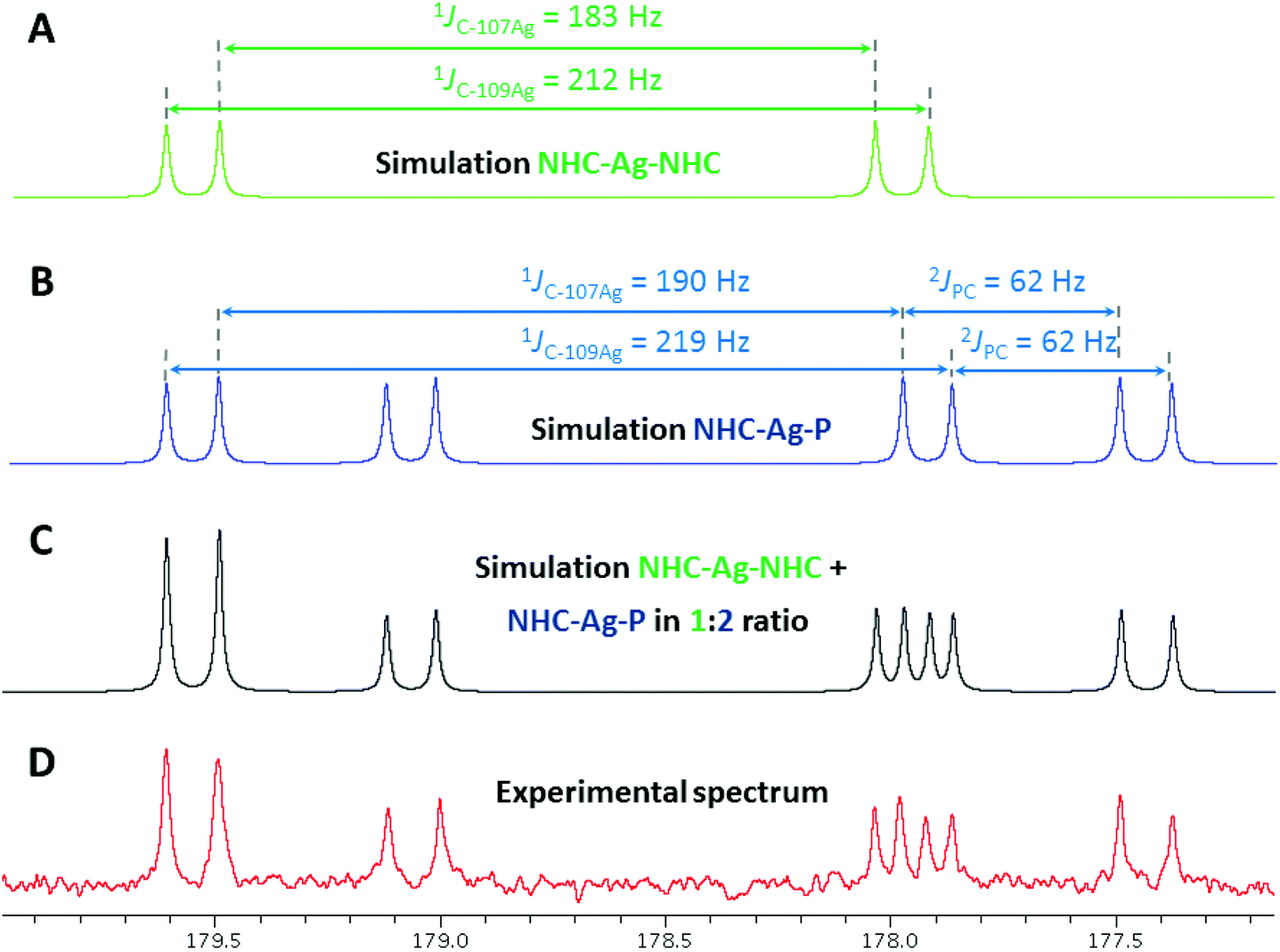 | ||
| Fig. 8 Details of the 13C{1H}-NMR spectrum of [Ag2(LtBu)2](BF4)2 in the δ 177–180 ppm region (D) and simulated spectra (TOPSPIN-DAISY module) of the homoleptic (HH) NHC–Ag–NHC isomer (A), the heteroleptic (HT) NHC–Ag–P isomer (B) and both the HH and HT isomers in a 1:2 ratio (C). Spectrum recorded in CD2Cl2 at 125.77 MHz and 298 K with a cryogenically cooled probe head; accumulation of 1024 scans. | ||
An X-ray diffraction study of [Ag2(LtBu)2](BF4)2 also revealed a ‘head-to-tail’ coordination of the bidentate ligand (Fig. 7, right), with two crystallographically independent but very similar dinuclear complexes in the unit cell (Table 1). The bond distances and angles in [Ag2(LR)2]2+ for R = Ph and tBu are very close or within experimental error, showing that the nature of the P donor group has only little influence on the solid state structure.
Interestingly, Hofmann and co-workers recently reported the formation of P–NHC-type ‘head-to-head’ and ‘head-to-tail’ copper(I) complexes.10f Depending on the nature of NHC wingtip, either the homoleptic or the heteroleptic isomer was isolated. Mutual ‘trans-coordination’ of the NHC and P donors, electronically disfavoured,37 was rationalised by minimisation of the steric repulsion in the ‘head-to-head’ complex. Yet for these complexes, no ‘head-to-head’/‘head-to-tail’ isomerisation was detected in different NMR solvents.
Synthesis and structure of dinuclear copper(I) complexes
We have already reported the synthesis of tetranuclear, ladder-type P–NHC-type CuI complexes by transmetallation from [Ag4Br4(LR)2] or by reaction of the phosphonium–imidazolium LR·2HBr salts with mesitylcopper(I) [Cu5(Mes)5],12e which has been used before to form CuI NHC complexes from imidazolium salts.38 The coordination chemistry of the LR ligands with CuI was further investigated by using the monoprotic pro-ligands LR·HBr.Reaction of LR·HBr (R = Ph, tBu, Cy) with [Cu5(Mes)5] resulted in the formation of the corresponding [Cu2Br2(LR)2] complexes in good yields (Scheme 4). Completion of the reaction was evidenced by 1H NMR spectroscopy (i.e. disappearance of the imidazolium NCHN signal). For all three CuI complexes, the 31P{1H}-NMR spectra revealed a singlet assignable to the coordinated P donor, only slightly shifted from the position observed in the starting LR·HBr. In the 13C{1H}-NMR spectra, the CuI–CNHC resonance was detected in the region δ 183–186 ppm, typical for CuI–NHCs.25b The CNHC signal was observed as a doublet (2JPC ≈ 46–47 Hz) for the dialkyl phosphine derivatives or as a broad signal for [Cu2Br2(LPh)2], possibly due to a different rate of fluxionality of the CNHC–Cu bonds in these two complexes. In the 1H-NMR spectrum of [Cu2Br2(LtBu)2], the line-shape of the signals for the methylene protons was field-dependent, pointing towards a dynamic process in solution.
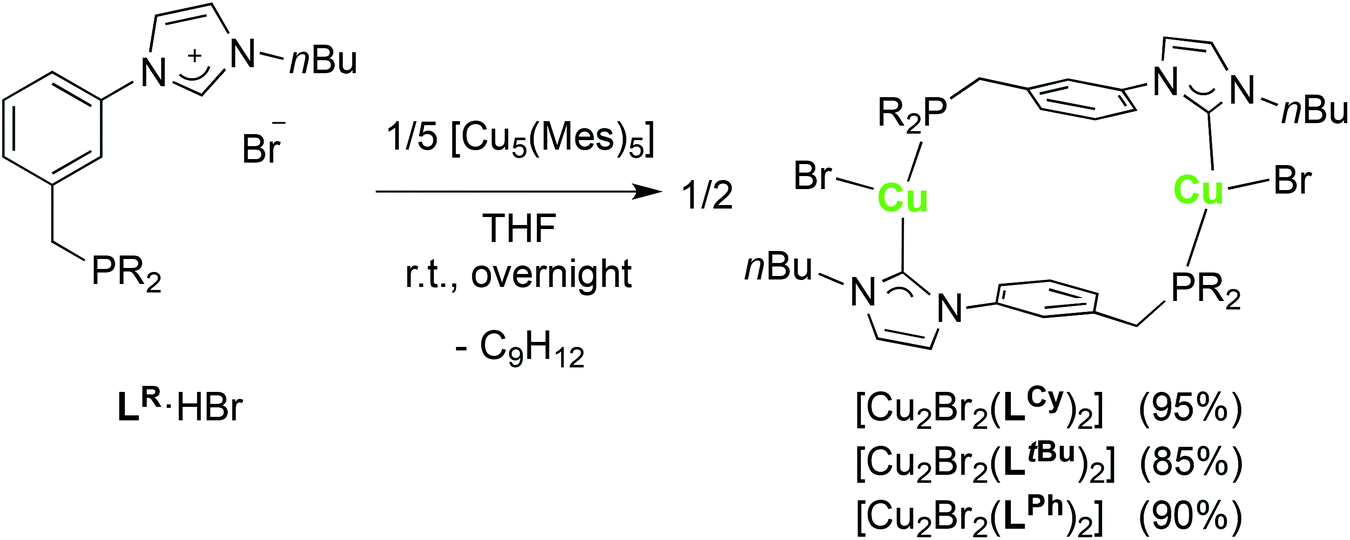 | ||
| Scheme 4 Synthesis of the dinuclear copper(I) complexes [Cu2Br2(LR)2]. Yields are based on LR. | ||
The structures of [Cu2Br2(LCy)2] and [Cu2Br2(LtBu)2]·2CH2Cl2 were determined crystallographically and are depicted in Fig. 9. Both complexes crystallised as dimers with two LR ligands bridging the two copper centres, reminiscent of the coordination behaviour of the ligands in [Ag2(LR)2](BF4)2. Both structures present a ‘head-to-tail’ arrangement for the NHC and P donors. The three-coordinate Cu centres adopt a distorted planar T-shaped coordination geometry, the third donor being a bromide. The Cu–CNHC distances, from 1.938(6) to 1.960(6) Å, and the Cu–P bond lengths lie within the range reported for related complexes.10f,39 The large separation between the two CuI centres (from 6.836(1) to 7.138(1) Å) can be traced to the large 1,3-phenylene spacer linking the NHC and phosphine donors.
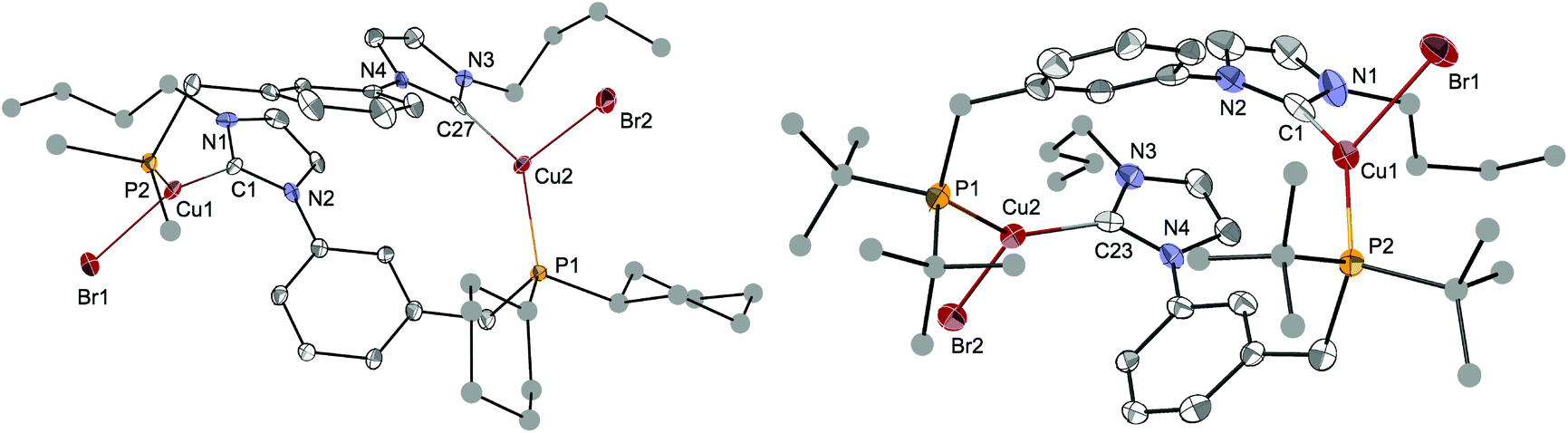 | ||
| Fig. 9 The molecular structures of [Cu2Br2(LCy)2] (left) and [Cu2Br2(LtBu)2]·2CH2Cl2 (right) with thermal ellipsoids at 40% probability. In [Cu2Br2(LCy)2] only one Cy carbon and one disordered position for the nBu chain are shown for clarity. C atoms for the nBu, Cy and tBu groups are depicted as spheres of arbitrary radii. H atoms and crystallisation solvents have been removed for clarity. Selected metrical data are given in Table 2. | ||
In order to study further the dynamic behaviour of the CuI complexes in solution, we undertook a variable temperature (VT) 1H-NMR study of [Cu2Br2(LtBu)2] in CD2Cl2 prompted by its relatively simple line-shape compared to the LPh and LCy analogues (Fig. 10). At room temperature, very broad signals were observed at 600 MHz for the various protons, suggesting possible coalescence. Upon cooling to −41 °C, two sharp doublets at δ 1.43 and 0.86 ppm (9 H each) assignable to the tBu groups on P indicated a static structure (HD). At this temperature, the signal of the methylene protons (HC) was split into two complex multiplets, due to the geminal 2JHH and 2JPH coupling in an ABX (A = B = H, X = P) spin system. Interestingly, the NCH2 protons (HB) of the NHC wingtip also appeared as diastereotopic. The backbone HA proton, closer to the aryl spacer, gives rise to a doublet at this temperature owing to 3JHH coupling. For comparison, at 35 °C, one broad singlet (18 H) was assignable to the tBu groups on P and a doublet was observed for the methylene protons (HC) in accordance with a relatively fast exchange of their positions on the NMR time scale. The spectral characteristics at lower temperature are in agreement with the solid-state structure being retained in solution. The dynamic behaviour at higher temperatures may have diverse origins, i.e. conformational changes in the dimeric structure involving flipping of the phenylene linker and/or the reversible formation of ‘head-to-head’ coordinated dimers by ligand (hemi)lability. The activation barrier corresponding to the fluxional behaviour of the tBu groups was found to be ΔG‡ = 56.5 ± 1.0 kJ mol−1. Based on the current data there is no preference for any of the above explanations. The latter hypothesis is however less likely since only one singlet is observed in the 31P{1H}-NMR spectrum at room temperature. Recent work involving ligands with NHC and P donors held together by a CH2 linker ascribed stereo-isomerisations at the Cu centre to fluxionality.10f
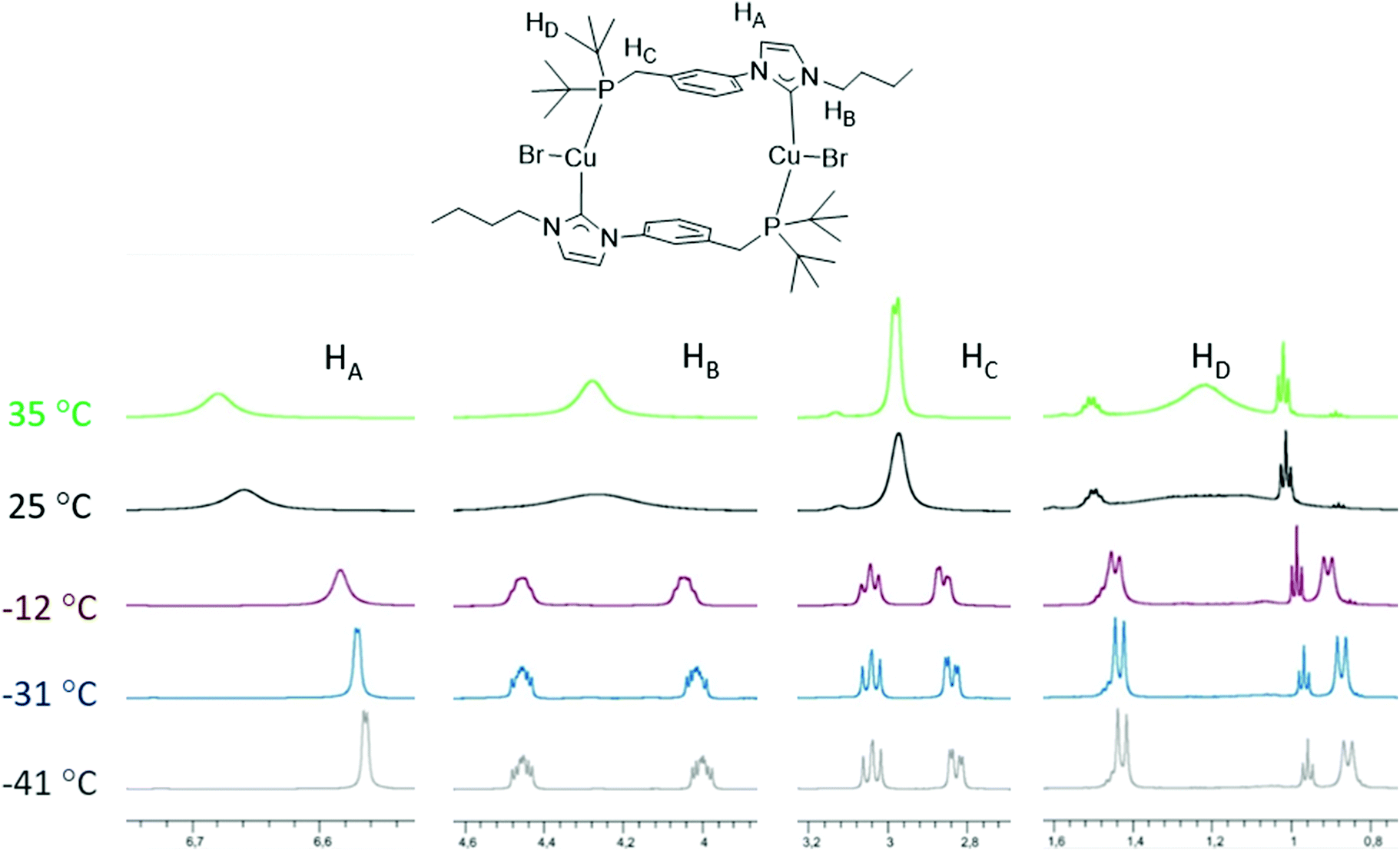 | ||
| Fig. 10 VT 1H NMR (600 MHz) spectra of [Cu2Br2(LtBu)2] in CD2Cl2 at temperatures from −41 to +35 °C. | ||
In contrast, the reaction of [Cu5(Mes)5] with the phosphonium–imidazolium LR·2HBr, or the transmetallation of the corresponding [Ag4Br4(LR)2] cubanes with 4 mole equiv. of [CuBr·SMe2] (R = Cy, tBu) gave rise to the tetranuclear clusters [Cu4Br4(LR)2].12e Metrical data regarding the di- and tetranuclear CuI complexes are reported in Table 2.
| [Cu2Br2(LtBu)2] | [Cu2Br2(LCy)2]b | [Cu4Br4(LCy)2]a | |
|---|---|---|---|
| a Data taken from ref. 12e. b There are two dinuclear complexes exhibiting similar metrical data in the asymmetric unit, the second set of values refers to the other molecule. | |||
| Cu1⋯Cu2 | 6.899(1) | 7.138(1)/6.836(1) | 2.790(1) |
| Cu1–Br1 | 2.497(1) | 2.453(1)/2.493(1) | 2.442(1) |
| Cu2–Br2 | 2.483(1) | 2.438(1)/2.493(1) | 2.503(1) |
| Cu1–P2 | 2.231(2) | 2.222(2)/2.230(2) | |
| Cu2–P1 | 2.237(2) | 2.217(2)/2.230(2) | 2.211(2) |
| Cu1–C1 | 1.960(6) | 1.952(6)/1.947(6) | 1.903(5) |
| Cu2–C23/C27 | 1.938(6) | 1.943(6)/1.947(6) | |
| P2–Cu1–Br1 | 114.8(1) | 109.7(1)/142.4(2) | |
| P2–Cu1–C1 | 145.1(2) | 137.9(2)/107.5(1) | |
| C1–Cu1–Br1 | 100.0(2) | 112.2(2)/110.1(2) | |
| ∑angles around Cu1 | 359.9 | 359.8/360.0 | |
| ∑angles around Cu2 | 360.0 | 360.0/360.0 | |
The longer Cu–CNHC and Cu–P bond distances in the [Cu2Br2(LR)2] complexes (mean distances ca. 1.948 and 2.228 Å, respectively) in comparison to the [Cu4Br4(LCy)2] cluster (1.903(5) Å and 2.211(2) Å) probably originate from the competition between mutually trans strong P and NHC σ-donors.
Synthesis and structure of a dinuclear gold(I) complex
Since transmetallation of the silver cubane [Ag4Br4(LtBu)2] with CuI always led to tetranuclear complexes,12e we wondered what would happen with AuI. Reaction of [Ag4Br4(LtBu)2] with 4 mole equiv. of [AuCl(THT)] led to the homodinuclear gold complex [Au2Cl2LtBu] (Scheme 5). 13C{1H}-NMR spectral analysis supported the NHC transmetallation as a downfield singlet was detected at δ 170.3 ppm, in a range typical for AuI–CNHC functionalities.40 A singlet at δ 79.0 ppm in the 31P{1H}-NMR spectrum also confirmed concomitant phosphine transfer to gold. However, a minor peak was observed at δ 80.1 ppm and ascribed to analogous complexes originating from partial halide scrambling (Cl/Br); this was also supported by elemental analysis (cf. Experimental section).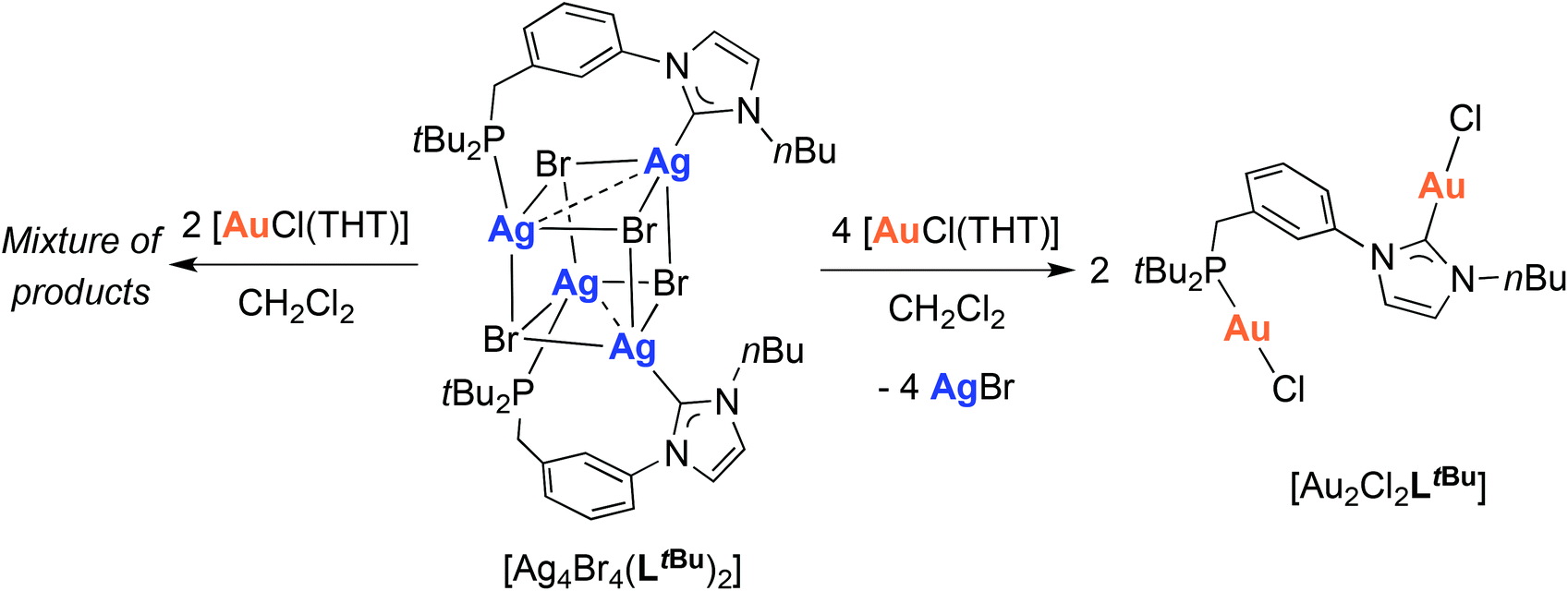 | ||
| Scheme 5 Transmetallation from [Ag4Br4(LtBu)2]12e to obtain the dinuclear [Au2Cl2LtBu] complex. | ||
The structure of [Au2Cl2LtBu] (Fig. 11) revealed an approximate linear coordination of the AuI centres (P–Au–Cl: 177.7(1)° and CNHC–Au–Cl: 176.4(2)°), common for NHC gold(I) complexes. The Au–CNHC (1.985(5) Å) and Au–P (2.239(1) Å) bond distances are in the expected range.19,40 Contrary to a recent report by Roesky and co-workers on related P–NHC-type gold(I) complexes (Fig. 3) obtained by transmetallation from the silver analogues,19 no intra- or inter-molecular Au–Au interactions were observed in the solid state for [Au2Cl2LtBu].
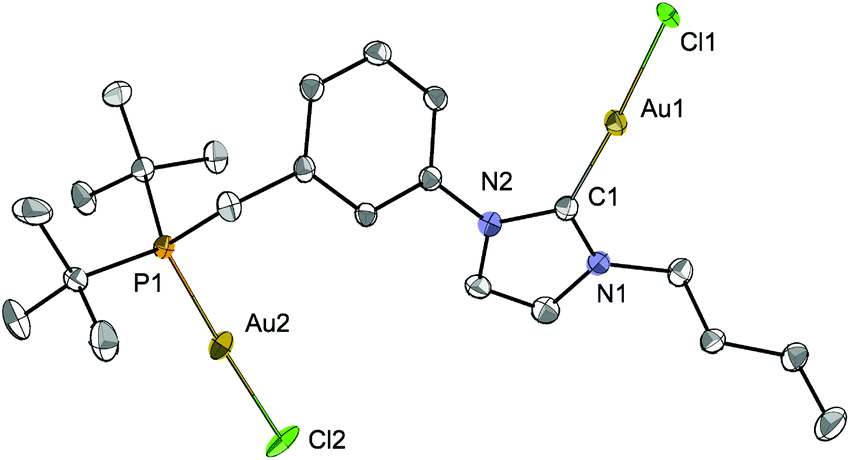 | ||
| Fig. 11 The molecular structure of [Au2Cl2LtBu] with thermal ellipsoids at 40% probability. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles [°]: Au1–Cl1 2.323(1), Au2–Cl2 2.325(1), Au1–C1 1.985(5), Au2–P1 2.239(1); C1–Au1–Cl1 176.39(15), P1–Au2–Cl2 177.69(5), N1–C1–N2 104.3(4). | ||
Attempts to synthesise heterobimetallic silver–gold complexes proved unsuccessful, as the reaction of [Ag4Br4(LtBu)2] with 2 mole equiv. of [AuCl(THT)] led to a mixture of products containing [Au2Cl2LtBu].
Conclusion
The rational synthesis of a range of hybrid P–NHC-type (pro-)ligands with systematically varied substitution at P, provided insight into their coordination chemistry with coinage metals. The main features observed can be summarised as follows: (i) in all cases studied, the ligands bridge two metal centres, irrespective of the type of phosphine donor; (ii) in the presence of Br−, all silver complexes isolated adopt structures based on the [Ag4Br4(LR)2] motif comprising a distorted Ag4Br4 cubane core, bridging LR ligands and weak metallophilic interactions; (iii) in the presence of the non-coordinating BF4−, [Ag2(LR)2](BF4)2 complexes were obtained with bridging ‘head-to-tail’ ligand arrangement in the solid state and ‘head-to-tail’/‘head-to-head’ isomerisation in solution; (iv) the nature of the R substituent on the P end does not impact the structures of the Ag complexes characterised, but seems to influence the rates of dynamic processes in solution, presumably due to competition of electronic and steric factors of the P donor. The relative lability of the two types of donor ends in P–NHC-type hybrid ligands has been inferred from the nature of products obtained from the reaction of [Ag4Br4(LR)2] with [Ir(COD)(μ-Cl)]2;12e (v) dinuclear [Cu2Br2(LR)2] complexes with bridging ligands were easily accessible from LR·HBr and [Cu5(Mes)5] and are also non-rigid in solution; (vi) transmetallation of [Ag4Br4(LR)2] with [AuCl(THT)] results in transfer of both donor groups of the hybrid P–NHC-type ligands, leading to the dinuclear [Au2Cl2LtBu] complex.Guided by the synthesis of non-symmetrical (pro)ligands and through the understanding of their emerging coordination chemistry, ligand alterations may be targeted to favour chelating and/or pincer rather than bridging coordination behaviour. In addition, the pre-organized tethering of the two types of strong σ-donors on the same skeleton (as on LR) will provide insight into the donor competition behaviour that may lead to (hemi)labile or stable complexes with catalytic potential.12e
Experimental section
General methods
All air- and moisture-sensitive manipulations were performed under dry argon atmosphere using standard Schlenk techniques. THF and Et2O were dried by refluxing over sodium/benzophenone ketyl and distilled under an argon atmosphere prior use. Methanol and ethanol were refluxed over CaH2, distilled under an argon atmosphere and stored over 3 Å molecular sieves. Other solvents (pentane, CH2Cl2, toluene and acetonitrile) were dried by passing through columns of activated alumina and subsequently purged with argon. C6D6 and toluene-d8 were distilled over KH; other deuterated solvents were dried over 4 Å (CD2Cl2 and CDCl3) or 3 Å (CD3OD) molecular sieves, degassed by freeze–pump–thaw cycles, and stored under argon. Mesityl copper(I)41 and [AuCl(THT)]42 were prepared according to literature methods and all other chemicals were obtained from commercial sources and used without further purification. The synthesis of 1-(3-(bromomethyl)phenyl)-3-butyl-1H-imidazol-3-ium bromide (A), LCy·2HBr, LtBu·2HBr, [Ag4Br4(LCy)2], [Ag4Br4(LtBu)2] and [Cu4Br4(LCy)2] has already been reported in a recent communication.12eNMR spectra were recorded on Bruker spectrometers (AVANCE I – 300 MHz, AVANCE III – 400 MHz, AVANCE III – 600 MHz or AVANCE I – 500 MHz equipped with a cryogenic probe). Downfield shifts are reported in ppm as positive and referenced using signals of the residual protio solvent (1H), the solvent (13C) or externally (31P, 11B). All NMR spectra were measured at 298 K, unless otherwise specified. The multiplicity of the signals is indicated as s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet and br = broad. Unequivocal determination of nJPC coupling constants in ambiguous cases was carried out by recording the 13C{1H}-NMR spectra on two different field spectrometers. Assignments (Fig. 12) were determined either on the basis of unambiguous chemical shifts, coupling patterns and 13C-DEPT experiments or 2D correlations (1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC). Spin-simulation was carried out using the DAISY module of the Topspin 2.1 software (BRUKER). Elemental analyses were performed by the “Service de microanalyses”, Université de Strasbourg. Electrospray mass spectra (ESI-MS) were recorded on a microTOF (Bruker Daltonics, Bremen, Germany) instrument using nitrogen as drying agent and nebulizing gas.
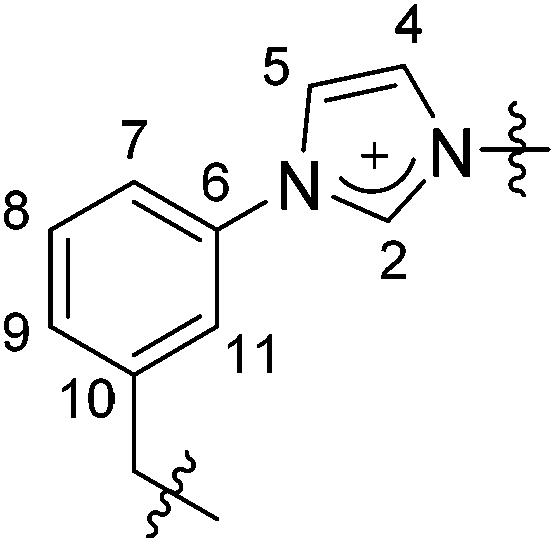 | ||
| Fig. 12 Atom numbering used for the assignment of the NMR resonances. | ||
General procedure for the synthesis of free carbene LR
To a suspension of LR·HBr (resp. LR·2HBr) in diethyl ether (10 mL), precooled at −78 °C, was added a solution of 1.1 equiv. (resp. 2.1 equiv.) of KN(SiMe3)2 in diethyl ether (10 mL). The resulting suspension was allowed to reach room temperature and stirred for 1 h, giving a suspension of KBr in a yellow solution. Removal of the volatiles under reduced pressure, extraction of the resulting oil with pentane (25 mL) filtration and evaporation of the solvent gave LR as a dark coloured oil.General procedure for the synthesis of silver(I) complexes [Ag2(LR)2](BF4)2
LR·HBF4 and Ag2O (0.55 equiv.) were charged in a Schlenk flask along with molecular sieves 4 Å. Degassed acetonitrile (15 mL) was added and the mixture was stirred for 2 days at 40 °C under exclusion of light. After evaporation of the solvent under reduced pressure, the remaining slurry was extracted twice with CH2Cl2 and the resulting solution was filtered over Celite® and concentrated to ca. 1 mL. The complex [Ag2(LR)2](BF4)2 was precipitated with diethyl ether. The white powder was collected by filtration and dried under vacuum.:1 HT/HH ratio in CD2Cl2 (see text) and 1:1.1 in CD3CN. 1H NMR (400.13 MHz, CD2Cl2): δ 8.00 (br q, 4JHH = 4JPH = 1.6 Hz, 0.8H, CHarom. H11), 7.64 (t, 3JHH = 4JHAg = 1.7 Hz, 0.8H, CHimid.), 7.63–7.22 (m, 11.6H, CHarom.), 7.17 (t, 3JHH = 4JHAg = 1.7 Hz, 0.8H, CHimid.), 7.09 (t, 3JHH = 7.9 Hz, 0.2H, CHarom. H8), 7.04 (t, 3JHH = 7.8 Hz, 0.8H, CHarom. H8), 6.92 (s, 0.2H, CHarom.), 6.60 (d, 3JHH = 7.7 Hz, CHarom. H7/H9), 4.30 (t, 3JHH = 7.3 Hz, 1.6H, NCH2), 4.20 (t, 3JHH = 7.3 Hz, 0.4H, NCH2), 3.93 (AB part of an ABX spin system with A = B = H and X = P, J = 10.5, 4.6 Hz, 1.6H, CH2P), 3.89–3.82 (br s, 0.4H, CH2P), 1.91 (quint, 3JHH = 7.4 Hz, 0.4H, NCH2CH2), 1.82 (quint, 3JHH = 7.4 Hz, 1.6H, NCH2CH2), 1.40 (sext, 3JHH = 7.4 Hz, 2H, NCH2CH2CH2), 1.03 (t, 3JHH = 7.3 Hz, 0.6H, CH3), 0.96 (t, 3JHH = 7.4 Hz, 2.4H, CH3). 13C{1H} NMR (125.77 MHz, CD2Cl2): δ CNHC peak not observed, 140.7 (Carom.), 137.6 (Carom.), 133.6 (dd, JPC = 14.6 Hz, JAgC = 2.3 Hz, CHPPh), 132.1 (d, JPC = 1.8 Hz, CHPPh), 130.4 (d, J = 5.4 Hz, CHarom.), 129.9 (d, J = 2.9 Hz, CHarom.), 129.8 (d, JPC = 10.4 Hz, CHPPh), 129.1 (dd, 1JPC = 36.7 Hz, 2JAgC = 4.4 Hz, CPPh), 126.3 (d, J = 8.4 Hz, CHarom.), 122.8 (d, J = 5.5 Hz, CHarom.), 122.7 (d, J = 4.0 Hz, CHarom.), 122.2 (d, J = 6.3 Hz, CHarom.), 52.3 (NCH2), 34.4 (d, 1JPC = 17.1 Hz, CH2P), 33.9 (NCH2CH2), 19.9 (NCH2CH2CH2), 13.9 (CH3). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 25.2 (two doublets, 1JP–107Ag = 503 Hz, 1JP–109Ag = 581 Hz, integrating for 0.8P), 17.6 (two doublets, 1JP–107Ag ≈ 515 Hz, 1JP–109Ag ≈ 595 Hz, integrating for 0.2P). 11B NMR (128.38 MHz, CD2Cl2): δ −0.9 (quint, 1JBF = 1.5 Hz).
1H NMR (400.13 MHz, CD3CN): δ 7.67–7.25 (m, 13.6H, CHarom.), 7.14–6.88 (br s, 2H, CHarom.), 6.82–6.60 (br s, 0.4H, CHarom.), 5.45 (s, 0.4H, residual CH2Cl2), 4.20 (t, 3JHH = 7.3 Hz, 2H, NCH2), 3.70 (br s, 2H, CH2P), 1.91–1.69 (br s, 2H, NCH2CH2), 1.37 (sext, 3JHH = 7.4 Hz, 2H, NCH2CH2CH2), 0.98 (t, 3JHH = 7.3 Hz, 3H, CH3). 31P{1H} NMR (161.98 MHz, CD3CN): δ 21.3 (two doublets, 1JP–107Ag ≈ 500 Hz, 1JP–109Ag ≈ 580 Hz, integrating for 1.0P), 11.2 (two doublets, 1JP–107Ag ≈ 475 Hz, 1JP–109Ag ≈ 550 Hz, integrating for 1.1P).
:2 in CD2Cl2 (see text). 1H NMR (400.13 MHz, CD2Cl2): 7.74 (br s, 0.7H, CHarom. H11), 7.66 (t, 3JHH = 4JHAg = 1.7 Hz, 0.7H, CHimid.), 7.58 (t, 3JHH = 4JHAg = 1.8 Hz, 0.3H, CHimid.), 7.50 (br s, 0.3H, CHarom. H11), 7.40 (br d, 3JHH = 7.8 Hz, 0.3H, CHarom. H7/H9), 7.35 (br d, 3JHH = 7.9 Hz, 0.7H, CHarom. H7/H9), 7.31 (t, 3JHH = 4JHAg = 1.8 Hz, 0.3H, CHimid.), 7.31–7.26 (overlapping m, 0.7H, CHarom. H9/H7), 7.28 (t, 3JHH = 4JHAg = 1.7 Hz, 0.7H, CHimid.), 7.23–7.19 (overlapping m, 0.3H, CHarom. H9/H7), 7.23 (t, 3JHH = 7.8 Hz, 0.7H, CHarom. H8), 7.14 (t, 3JHH = 7.8 Hz, 0.3H, CHarom. H8), 4.25 (t, 3JHH = 7.3 Hz, 0.6H, NCH2), 4.18 (t, 3JHH = 7.3 Hz, 1.4H, NCH2), 3.32 (AB part of an ABX spin system with A = B = H and X = P, J = 10.1 Hz, 4.2 Hz, 1.4H, CH2P), 3.27 (AB part of an ABX spin system with A = B = H and X = P, J = 9.0 Hz, 3.7 Hz, 0.6H, CH2P), 1.96 (quint, 3JHH = 7.5 Hz, 0.6H, NCH2CH2), 1.86 (quint, 3JHH = 7.5 Hz, 1.4H, NCH2CH2), 1.46 (sext, 3JHH = 7.5 Hz, 2H, NCH2CH2CH2), 1.40 (virtual t, |3JPCH + 5JPH| = 14.9 Hz, 6H, C(CH3)3), 1.29 (d, 3JPH = 14.7 Hz, 12H, C(CH3)3), 1.04 (t, 3JHH = 7.3 Hz, 1H, CH3), 1.01 (t, 3JHH = 7.4 Hz, 2H, CH3). 13C{1H} NMR (125.77 MHz, CD2Cl2): δ 178.8 (two doublets, 1JC–107Ag = 183 Hz, 1JC–109Ag = 212 Hz, minor CNHC), 178.5 (two dd, 1JC–107Ag = 190 Hz, 1JC–109Ag = 219 Hz, 2JP–Ag–C = 62 Hz, major CNHC), 141.3 (minor Carom.), 141.1 (major Carom.), 140.6 (minor Carom.), 140.2 (major Carom.), 131.1 (d, JPC = 5.7 Hz, major CHarom.), 130.7 (minor CHimid.), 130.3 (major CHimid.), 129.1 (virtual t, |nJPC + n+2JPAgPC| = 8.2 Hz, minor CHarom.), 125.9 (d, JPC = 9.6 Hz, major CHarom.), 125.2 (virtual t, |nJPC + n+2JPAgPC| = 6.0 Hz, minor CHarom.), 123.0 (d, JPC = 5.8 Hz, major CHarom.), 122.9 (d, JPC = 5.8 Hz, minor CHarom.), 122.8 (d, J = 5.2 Hz, minor CHarom.), 122.5 (minor CHimid.), 122.3 (major CHimid.), 122.1 (d, J = 5.3 Hz, major CHarom.), 52.7 (minor NCH2), 52.6 (major NCH2), 35.3 (doublet of virtual t, |1JPC + 3JPAgPC| = 11.9 Hz, 2JCAg = 3.9 Hz, minor C(CH3)3), 34.8 (dd, 1JPC = 12.2 Hz, 2JCAg = 3.7 Hz, major C(CH3)3), 34.3 (major NCH2CH2), 33.9 (minor NCH2CH2), 29.95–29.75 (m, C(CH3)3), 27.0 (d, 1JPC = 12.9 Hz, major CH2P), 26.4 (virtual t, |1JPC + 3JPAgPC| = 14.0 Hz, minor CH2P), 20.3 (minor NCH2CH2CH2), 20.1 (major NCH2CH2CH2), 13.84 (major CH3), 13.81 (minor CH3). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 73.6 (two doublets, 1JP–107Ag = 460 Hz, 1JP–109Ag = 530 Hz, integrating for 1P), 72.4 (two doublets, 1JP–107Ag = 477 Hz, 1JP–109Ag = 552 Hz, integrating for 2P).
General procedure for the synthesis of copper(I) complexes [Cu2Br2(LR)2]
To a suspension of LR·HBr in THF (5 mL) was added a solution of mesityl copper(I) (1.0 equiv. based on Cu) in THF (5 mL) at room temperature. The resulting clear solution was stirred at room temperature for 24 h. After evaporation of the solvent under reduced pressure, the solid residue was washed with diethyl ether and redissolved in CH2Cl2. Addition of pentane precipitated [Cu2Br2(LR)2] as a white powder that was collected and dried under vacuum.X-ray crystallography
Suitable crystals for the X-ray analysis of all compounds were obtained as described above. Summary of the crystal data, data collection and refinement are given in Table S1 (see ESI‡).Data sets for LPh·HBr, LCy·HBr·BH3, [Ag4Br4(LPh)2], [Ag2(LPh)2](BF4)2·2CH2Cl2, [Cu2Br2(LCy)2] and [Au2Cl2LtBu] were collected at 173(2) K on a Bruker APEX-II CCD Duo diffractometer (graphite-monochromated Mo-Kα radiation, λ = 0.71073 Å). Data sets for [Ag2(LPh)2](BF4)2, [Ag2(LtBu)2](BF4)2·CH2Cl2 and [Cu2Br2(LtBu)2]·2CH2Cl2 were collected at 173(2) K on a Kappa CCD diffractometer (graphite-monochromated Mo-Kα radiation, λ = 0.71073 Å). Specific comments for each data set are given below. The cell parameters were determined using DENZO43 (Kappa) or APEX244 (APEX-II) softwares. The structures were solved by direct methods using the program SHELXS-97 (compounds LCy·HBr·BH3, LPh·HBr, [Cu2Br2(LCy)2] and [Cu2Br2(LtBu)2]·2CH2Cl2) or SHELXS-2013 (complexes [Ag4Br4(LPh)2], [Ag2(LPh)2](BF4)2·2CH2Cl2, [Ag2(LPh)2](BF4)2, [Ag2(LtBu)2](BF4)2·CH2Cl2 and [Au2Cl2LtBu]).45 The refinement and all further calculations were carried out using SHELXL-97 (compound LPh·HBr, [Cu2Br2(LCy)2] and [Cu2Br2(LtBu)2]·2CH2Cl2) or SHELXL-2013 (all other compounds).45b The H-atoms were introduced into the geometrically calculated positions (SHELXL-97 or SHELXL-2013 procedures) unless stated otherwise and refined riding on the corresponding parent atoms. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2.
The following special comments apply to the models of the structures:
LCy·HBr·BH3: the alkyl atoms C5, C6 and C7 are disordered on two positions.
LPh·HBr: A SQUEEZE procedure46 was applied and the residual electron density was assigned to one half disordered molecule of CH2Cl2.
[Ag4Br4(LPh)2]: A SQUEEZE procedure46 was applied and the residual electron density was assigned to one disordered molecule of ether.
[Ag2(LPh)2](BF4)2: the alkyl atoms C31, C32 and C33 are disordered on two positions. A SQUEEZE procedure46 was applied and the residual electron density was assigned to two disordered molecules of acetonitrile. The structure of this complex can be found in the ESI.‡
[Ag2(LtBu)2](BF4)2·CH2Cl2: thermal motions affect the alkyl chains on the ligands. The carbons atoms C49, C50 and C73 are disordered on two positions. The carbon atom C48 is also disordered on two positions but C48 and C48B have been imposed at the same position to avoid short-contacts between the H-atoms and subsequent alerts in the Checkcif. A SQUEEZE procedure46 was applied and the residual electron density was assigned to one and a half disordered molecules of CH2Cl2.
[Cu2Br2(LCy)2]: The asymmetric unit contains one and a half molecules of the complex. The alkyl atoms C6 and C7 are disordered on two positions.
[Cu2Br2(LtBu)2]·2CH2Cl2: The space group is chiral (P21) and the value of Flack parameter is −0.008(9). A SQUEEZE procedure46 was applied and the residual electron density was assigned to one disordered molecule of toluene.
Acknowledgements
The USIAS, CNRS, Unistra, Région Alsace and Communauté Urbaine de Strasbourg are acknowledged for the award of fellowships and a Gutenberg Excellence Chair (2010–11) to AAD. We thank the CNRS and the MESR (Paris) for funding and for a PhD grant to TS. We are grateful to Corinne Bailly and Dr Lydia Karmazin (Service de Radiocristallographie, Unistra) for solving crystal structures, and to Maurice Coppe and Dr Bruno Vincent (Service de Résonance Magnétique Nucléaire, Unistra) for assistance in the NMR studies. We thank Johnson Matthey PLC for a generous loan of gold precursors.References
- (a) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS; (b) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407–5413 CrossRef CAS; (c) R. H. Crabtree, J. Organomet. Chem., 2005, 690, 5451–5457 CrossRef CAS.
- (a) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS; (b) J. C. Green, R. G. Scurr, P. L. Arnold and F. G. N. Cloke, Chem. Commun., 1997, 1963–1964 RSC; (c) C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801–5809 CrossRef CAS; (d) M.-T. Lee and C.-H. Hu, Organometallics, 2004, 23, 976–983 CrossRef CAS; (e) R. Tonner, G. Heydenrych and G. Frenking, Chem. – Asian J., 2007, 2, 1555–1567 CrossRef CAS PubMed; (f) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS; (g) A. Comas-Vives and J. N. Harvey, Eur. J. Inorg. Chem., 2011, 5025–5035 CrossRef CAS; (h) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- (a) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS PubMed; (b) W. A. Herrmann, V. P. W. Böhm, C. W. K. Gstöttmayr, M. Grosche, C.-P. Reisinger and T. Weskamp, J. Organomet. Chem., 2001, 617–618, 616–628 CrossRef CAS; (c) G. D. Frey, J. Schütz, E. Herdtweck and W. A. Herrmann, Organometallics, 2005, 24, 4416–4426 CrossRef CAS; (d) A. Flahaut, S. Roland and P. Mangeney, J. Organomet. Chem., 2007, 692, 5754–5762 CrossRef CAS; (e) V. Jurčík, S. P. Nolan and C. S. J. Cazin, Chem. – Eur. J., 2009, 15, 2509–2511 CrossRef PubMed; (f) T. E. Schmid, D. C. Jones, O. Songis, O. Diebolt, M. R. L. Furst, A. M. Z. Slawin and C. S. J. Cazin, Dalton Trans., 2013, 42, 7345–7353 RSC; (g) J. Broggi, V. Jurčík, O. Songis, A. Poater, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, J. Am. Chem. Soc., 2013, 135, 4588–4591 CrossRef CAS PubMed; (h) K. Farrell, H. Müller-Bunz and M. Albrecht, Organometallics, 2015, 34, 5723–5733 CrossRef CAS.
- (a) F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969–7975 CrossRef CAS; (b) S. Guo, M. H. Lim and H. V. Huynh, Organometallics, 2013, 32, 7225–7233 CrossRef CAS.
- (a) P. G. Edwards and F. E. Hahn, Dalton Trans., 2011, 40, 10278–10288 RSC; (b) S. Gaillard and J.-L. Renaud, Dalton Trans., 2013, 42, 7255–7270 RSC; (c) P. Hofmann and M. Brill, in Molecular Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 207–234 Search PubMed.
- (a) C. Yang, H. M. Lee and S. P. Nolan, Org. Lett., 2001, 3, 1511–1514 CrossRef CAS PubMed; (b) N. Tsoureas, A. A. Danopoulos, A. A. D. Tulloch and M. E. Light, Organometallics, 2003, 22, 4750–4758 CrossRef CAS; (c) H. M. Lee, P. L. Chiu and J. Y. Zeng, Inorg. Chim. Acta, 2004, 357, 4313–4321 CrossRef CAS; (d) A.-E. Wang, J.-H. Xie, L.-X. Wang and Q.-L. Zhou, Tetrahedron, 2005, 61, 259–266 CrossRef CAS; (e) R. Hodgson and R. E. Douthwaite, J. Organomet. Chem., 2005, 690, 5822–5831 CrossRef CAS; (f) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2006, 25, 5927–5936 CrossRef CAS; (g) J. Zhong, J.-H. Xie, A.-E. Wang, W. Zhang and Q.-L. Zhou, Synlett, 2006, 1193–1196 CAS; (h) X. Quan, S. Kerdphon and P. G. Andersson, Chem. – Eur. J., 2015, 21, 3576–3579 CrossRef CAS PubMed.
- C. A. Wheaton, J.-P. J. Bow and M. Stradiotto, Organometallics, 2013, 32, 6148–6161 CrossRef CAS.
- J. Witt, A. Pöthig, F. E. Kühn and W. Baratta, Organometallics, 2013, 32, 4042–4045 CrossRef CAS.
- (a) A. P. Marchenko, H. N. Koidan, A. N. Hurieva, O. V. Gutov, A. N. Kostyuk, C. Tubaro, S. Lollo, A. Lanza, F. Nestola and A. Biffis, Organometallics, 2013, 32, 718–721 CrossRef CAS; (b) P. Nägele, U. Herrlich, F. Rominger and P. Hofmann, Organometallics, 2013, 32, 181–191 CrossRef; (c) P. Ai, A. A. Danopoulos, P. Braunstein and K. Yu Monakhov, Chem. Commun., 2014, 50, 103–105 RSC; (d) C. C. Brown, F. Rominger, M. Limbach and P. Hofmann, Inorg. Chem., 2015, 54, 10126–10140 CrossRef CAS PubMed; (e) P. Ai, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2015, 54, 3722–3724 CrossRef CAS PubMed.
- (a) W. A. Herrmann, C. Köcher, L. J. Gooßen and G. R. J. Artus, Chem. – Eur. J., 1996, 2, 1627–1636 CrossRef CAS; (b) A. A. Danopoulos, S. Winston, T. Gelbrich, M. B. Hursthouse and R. P. Tooze, Chem. Commun., 2002, 482–483 RSC; (c) J. Wolf, A. Labande, J.-C. Daran and R. Poli, J. Organomet. Chem., 2006, 691, 433–443 CrossRef CAS; (d) J. Wolf, A. Labande, J.-C. Daran and R. Poli, Eur. J. Inorg. Chem., 2008, 3024–3030 CrossRef CAS; (e) G. Song, X. Li, Z. Song, J. Zhao and H. Zhang, Chem. – Eur. J., 2009, 15, 5535–5544 CrossRef CAS PubMed; (f) E. Kühnel, I. V. Shishkov, F. Rominger, T. Oeser and P. Hofmann, Organometallics, 2012, 31, 8000–8011 CrossRef; (g) M. Brill, E. Kühnel, C. Scriban, F. Rominger and P. Hofmann, Dalton Trans., 2013, 42, 12861–12864 RSC; (h) M. Brill, D. Marrwitz, F. Rominger and P. Hofmann, J. Organomet. Chem., 2015, 775, 137–151 CrossRef CAS.
- (a) E. Bappert and G. Helmchen, Synlett, 2004, 1789–1793 CAS; (b) C.-C. Ho, S. Chatterjee, T.-L. Wu, K.-T. Chan, Y.-W. Chang, T.-H. Hsiao and H. M. Lee, Organometallics, 2009, 28, 2837–2847 CrossRef CAS; (c) I. Abdellah, Y. Canac, C. D. Mboyi, C. Duhayon and R. Chauvin, J. Organomet. Chem., 2015, 776, 149–152 CrossRef CAS.
- (a) M. Raynal, X. Liu, R. Pattacini, C. Vallée, H. Olivier-Bourbigou and P. Braunstein, Dalton Trans., 2009, 7288–7293 RSC; (b) B. Vabre, Y. Canac, C. Duhayon, R. Chauvin and D. Zargarian, Chem. Commun., 2012, 48, 10446–10448 RSC; (c) X. Liu and P. Braunstein, Inorg. Chem., 2013, 52, 7367–7379 CrossRef CAS PubMed; (d) B. Vabre, Y. Canac, C. Lepetit, C. Duhayon, R. Chauvin and D. Zargarian, Chem. – Eur. J., 2015, 21, 17403–17414 CrossRef CAS PubMed; (e) T. Simler, P. Braunstein and A. A. Danopoulos, Angew. Chem., Int. Ed., 2015, 54, 13691–13695 CrossRef CAS PubMed.
- (a) H. M. Lee, J. Y. Zeng, C.-H. Hu and M.-T. Lee, Inorg. Chem., 2004, 43, 6822–6829 CrossRef CAS PubMed; (b) S. Gischig and A. Togni, Organometallics, 2005, 24, 203–205 CrossRef CAS; (c) T. Steinke, B. K. Shaw, H. Jong, B. O. Patrick and M. D. Fryzuk, Organometallics, 2009, 28, 2830–2836 CrossRef CAS; (d) D. A. Valyaev, O. A. Filippov, N. Lugan, G. Lavigne and N. A. Ustynyuk, Angew. Chem., Int. Ed., 2015, 54, 6315–6319 CrossRef CAS PubMed.
- The Cambridge Structural Database, accessed Jan. 2016: F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- (a) P. L. Chiu and H. M. Lee, Organometallics, 2005, 24, 1692–1702 CrossRef CAS; (b) H. Salem, M. Schmitt, U. Herrlich, E. Kühnel, M. Brill, P. Nägele, A. L. Bogado, F. Rominger and P. Hofmann, Organometallics, 2013, 32, 29–46 CrossRef CAS; (c) A. Marchenko, H. Koidan, A. Hurieva, O. Kurpiieva, Y. Vlasenko, A. Kostyuk, C. Tubaro, A. Lenarda, A. Biffis and C. Graiff, J. Organomet. Chem., 2014, 771, 14–23 CrossRef CAS.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- M. E. Humphries, W. H. Pecak, S. A. Hohenboken, S. R. Alvarado, D. C. Swenson and G. J. Domski, Inorg. Chem. Commun., 2013, 37, 138–143 CrossRef CAS.
- J. Y. Zeng, M.-H. Hsieh and H. M. Lee, J. Organomet. Chem., 2005, 690, 5662–5671 CrossRef CAS.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2015, 21, 601–614 CrossRef CAS PubMed.
- D. Hérault, D. H. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508–2528 RSC.
- (a) A. A. Danopoulos, N. Tsoureas, S. A. Macgregor and C. Smith, Organometallics, 2007, 26, 253–263 CrossRef CAS; (b) G. Song, X. Wang, Y. Li and X. Li, Organometallics, 2008, 27, 1187–1192 CrossRef CAS.
- (a) T. Steiner, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 456–463 CrossRef; (b) T. Steiner and G. R. Desiraju, Chem. Commun., 1998, 891–892 RSC; (c) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS; (d) A. Kovács and Z. Varga, Coord. Chem. Rev., 2006, 250, 710–727 CrossRef.
- V. Charra, P. de Frémont, P.-A. R. Breuil, H. Olivier-Bourbigou and P. Braunstein, J. Organomet. Chem., 2015, 795, 25–33 CrossRef CAS.
- D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385–3407 CrossRef CAS PubMed.
- (a) J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed; (b) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed.
- W. D. Clark, G. E. Tyson, T. K. Hollis, H. U. Valle, E. J. Valente, A. G. Oliver and M. P. Dukes, Dalton Trans., 2013, 42, 7338–7344 RSC.
- X. Hu, I. Castro-Rodriguez, K. Olsen and K. Meyer, Organometallics, 2004, 23, 755–764 CrossRef CAS.
- H.-L. Su, L. M. Pérez, S.-J. Lee, J. H. Reibenspies, H. S. Bazzi and D. E. Bergbreiter, Organometallics, 2012, 31, 4063–4071 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC; (b) H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- (a) B.-K. Teo and J. C. Calabrese, J. Chem. Soc., Chem. Commun., 1976, 185–186 RSC; (b) G. J. S. Venter, A. Roodt and R. Meijboom, Inorg. Chim. Acta, 2009, 362, 2475–2479 CrossRef CAS.
- T. A. P. Paulose, S.-C. Wu, J. A. Olson, T. Chau, N. Theaker, M. Hassler, J. W. Quail and S. R. Foley, Dalton Trans., 2012, 41, 251–260 RSC.
- B.-K. Teo and J. C. Calabrese, Inorg. Chem., 1976, 15, 2467–2474 CrossRef CAS.
- (a) D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2002, 327, 116–125 CrossRef CAS; (b) D. Qin, X. Zeng, Q. Li, F. Xu, H. Song and Z.-Z. Zhang, Chem. Commun., 2007, 147–149 RSC; (c) F. Jean-Baptiste dit Dominique, H. Gornitzka and C. Hemmert, J. Organomet. Chem., 2008, 693, 579–583 CrossRef CAS; (d) C. E. Willans, K. M. Anderson, M. J. Paterson, P. C. Junk, L. J. Barbour and J. W. Steed, Eur. J. Inorg. Chem., 2009, 2835–2843 CrossRef CAS; (e) A. Rit, T. Pape and F. E. Hahn, Organometallics, 2011, 30, 6393–6401 CrossRef CAS.
- (a) J. M. Jenkins and B. L. Shaw, J. Chem. Soc. A, 1966, 770–775 RSC; (b) A. Pidcock, Chem. Commun., 1968, 92–92 RSC.
- (a) A. J. Arduengo III, H. V. R. Dias, J. C. Calabrese and F. Davidson, Organometallics, 1993, 12, 3405–3409 CrossRef; (b) B. Bildstein, M. Malaun, H. Kopacka, K. Wurst, M. Mitterböck, K.-H. Ongania, G. Opromolla and P. Zanello, Organometallics, 1999, 18, 4325–4336 CrossRef CAS; (c) S. Hameury, P. de Frémont, P.-A. R. Breuil, H. Olivier-Bourbigou and P. Braunstein, Dalton Trans., 2014, 43, 4700–4710 RSC.
- (a) R. G. Pearson, Inorg. Chem., 1973, 12, 712–713 CrossRef CAS; (b) S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885–9895 CrossRef CAS PubMed.
- A. A. Danopoulos, P. Cole, S. P. Downing and D. Pugh, J. Organomet. Chem., 2008, 693, 3369–3374 CrossRef CAS.
- (a) A. A. D. Tulloch, A. A. Danopoulos, S. Kleinhenz, M. E. Light, M. B. Hursthouse and G. Eastham, Organometallics, 2001, 20, 2027–2031 CrossRef CAS; (b) N. Schneider, V. César, S. Bellemin-Laponnaz and L. H. Gade, J. Organomet. Chem., 2005, 690, 5556–5561 CrossRef CAS; (c) J. Broggi, S. Díez-González, J. L. Petersen, S. Berteina-Raboin, S. P. Nolan and L. A. Agrofoglio, Synthesis, 2008, 141–148 CAS; (d) I. V. Shishkov, F. Rominger and P. Hofmann, Dalton Trans., 2009, 1428–1435 RSC; (e) S. Simonovic, A. C. Whitwood, W. Clegg, R. W. Harrington, M. B. Hursthouse, L. Male and R. E. Douthwaite, Eur. J. Inorg. Chem., 2009, 1786–1795 CrossRef CAS; (f) M. Bessel, F. Rominger and B. F. Straub, Synthesis, 2010, 1459–1466 CAS; (g) S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC; (h) M. Slivarichova, R. Ahmad, Y.-Y. Kuo, J. Nunn, M. F. Haddow, H. Othman and G. R. Owen, Organometallics, 2011, 30, 4779–4787 CrossRef CAS; (i) C. E. Strasser and V. J. Catalano, Inorg. Chem., 2011, 50, 11228–11234 CrossRef CAS PubMed; (j) B. R. M. Lake, E. K. Bullough, T. J. Williams, A. C. Whitwood, M. A. Little and C. E. Willans, Chem. Commun., 2012, 48, 4887–4889 RSC; (k) B. Liu, C. Chen, Y. Zhang, X. Liu and W. Chen, Organometallics, 2013, 32, 5451–5460 CrossRef CAS; (l) B. R. M. Lake and C. E. Willans, Chem. – Eur. J., 2013, 19, 16780–16790 CrossRef CAS PubMed.
- P. de Frémont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef.
- (a) S. Gambarotta, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Chem. Commun., 1983, 1156–1158 RSC; (b) M. Stollenz and F. Meyer, Organometallics, 2012, 31, 7708–7727 CrossRef CAS.
- R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, in Inorganic Syntheses, John Wiley & Sons, Inc., 2007, vol. 26, pp. 85–91 Search PubMed.
- R. W. W. Hooft, DENZO and COLLECT, Nonius BV, Delft, The Netherlands, 1998 Search PubMed.
- APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, SHELX2013, University of Göttingen, Germany, 2013 Search PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Prof. Peter Hofmann, a dear colleague and friend who made major research advancements and contributed much to the promotion of chemistry. |
| ‡ Electronic supplementary information (ESI) available: X-ray structure of [Ag2(LPh)2](BF4)2 and crystallographic summary table. CCDC 1445698–1445706. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00275g |
| This journal is © The Royal Society of Chemistry 2016 |