
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium nanoparticles in catalytic carbon nanoreactors: the effect of confinement on Suzuki–Miyaura reactions†
B.
Cornelio
ab,
A. R.
Saunders
c,
W. A.
Solomonsz
c,
M.
Laronze-Cochard
b,
A.
Fontana
a,
J.
Sapi
b,
A. N.
Khlobystov
cd and
G. A.
Rance
*c
aDipartimento di Farmacia, Università G. d'Annunzio, Via dei Vestini, I-66100 Chieti, Italy
bInstitut de Chimie Moléculaire de Reims, CNRS UMR 7312, Université de Reims Champagne-Ardenne, UFR Pharmacie, 51 Rue Cognacq-Jay, F-51096 Reims Cedex, France
cSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: graham.rance@nottingham.ac.uk; Fax: +44 115 9513563
dNottingham Nanotechnology and Nanoscience Centre, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
First published on 6th January 2015
Abstract
We explore the construction and performance of a range of catalytic nanoreactors based on palladium nanoparticles encapsulated in hollow graphitised nanofibres. The optimum catalytic material, with small palladium nanoparticles located almost exclusively at the graphitic step-edges within nanoreactors, exhibits attractive catalytic properties in Suzuki–Miyaura cross-coupling reactions. Confinement of nanoparticles at the step-edges facilitates retention of catalytic centres and recycling of catalytic nanoreactors without any significant loss of activity or selectivity over multiple catalytic cycles. Furthermore, careful comparison of the catalytic properties of palladium nanoparticles either on or in nanoreactors reveals that nanoscale confinement of catalysts fundamentally affects the pathways of the Suzuki–Miyaura reaction, with the yield and selectivity for the cross-coupled product critically dependent on the steric properties of the aryl iodide reactant, whereas no effects of confinement are observed for aryl boronic acid reactants possessing substituents in different positions. These results indicate that the oxidative addition step of the Suzuki–Miyaura reaction occurs at the step-edge of nanofibres, where the mechanisms and kinetics of chemical reactions are known to be sensitive to nanoscale confinement, and thus the extent of confinement in carbon nanoreactors can be discretely controlled by careful selection of the aryl iodide reactant.
Introduction
The confinement of chemical reactions inside carbon nanotubes (CNT) is emerging as a highly versatile tool to control the pathways of molecular transformations and is leading to the formation of new molecules and materials inaccessible by other means.1–4 Although the use of molecular species, such as cyclodextrins, calixarenes, cucurbiturils, supramolecular and coordination cages, micelles and bilayer vesicles,5–10 and extended porous solids, including zeolites,6,11 to hold chemical reactions has been well established over the last three decades, carbon nanotubes and related hollow carbon nanostructures possess superior chemical,12 thermal13 and mechanical stability13,14 and rely solely upon ubiquitous van der Waals interactions to facilitate transport of the reactants into the internal channel.15–21 For these reasons, they have the potential to outperform previously studied hollow nanoscale materials and permit exploration of new chemical processes.15,21–23 In our recent studies, we have evaluated the properties of catalytic chemical reactions confined within hollow graphitised nanofibres (GNF).24–27 This unique carbon nanomaterial possesses a topologically complex nanoscale structure comprising a series of stacked cups encased within concentric tubes. The internal diameters are typically between 50 and 70 nm, an order of magnitude greater than the dimensions of simple molecular species, and the nanofibre tips are always open, thus allowing efficient transport of molecules through the internal structure. Furthermore, the distinctive internal herringbone structure results in a succession of 3–4 nm high steps formed by rolled-up sheets of graphene that provide effective “anchoring points” for molecules and nanoparticles.28 This creates localised nanoscale reaction environments, different to the bulk phase, where the local concentration of aromatic reactants is maximised through attractive non-covalent interactions between the reactant and the underlying graphitic surface of the nanoreactor24–27 and the activity,25,27 selectivity24,26,27 and stability25 of confined metal nanoparticle catalysts are enhanced across a range of preparative chemical transformations. Thus, GNF represents the ideal nanoscale container to further probe the effects of confinement on chemical reactions.In this study, we explore the effect of confinement in GNF on the palladium-catalysed Suzuki–Miyaura reaction.29 This versatile cross-coupling reaction represents one of the most powerful carbon–carbon bond forming methodologies30,31 and is applicable to the broadest spectrum of possible substrates.32 Today, it is one of the most influential methods utilised in the synthesis of natural products and pharmaceuticals,33–35 with its importance recognised by the award of the 2010 Nobel Prize in Chemistry to its pioneering discoverer.36 However, despite the vast number of studies probing the influence of both molecular and nanoparticulate catalysts on the mechanism and properties of the Suzuki–Miyaura reaction,31,37–40 to the best of our knowledge only a small number of studies have probed the influence of nanoscale confinement on this important cross-coupling reaction.41–45 Often conducted within the pores of mesoporous silica,41–44 the confined reactions catalysed by palladium nanoparticles (PdNP) often proceed with the attractive catalytic properties of near selective formation in high yields of the cross-coupled product. Yet, as no comparison to the same catalytic species located on the exterior surface of the support material was possible, the sole effect of confinement on the properties of both the catalyst and the reaction could not be disclosed. Through systematic investigation of the assembly and catalytic properties of a range of different palladium nanoparticle–graphitised nanofibre composites, we have procured suitable materials that allow us to definitively discriminate between the effects of support and confinement. Our ideal catalytic nanoreactor, comprising small palladium catalytic centres located nearly exclusively at the internal step-edges within GNF, exhibits relatively good activity, selectivity and stability in the carbon–carbon bond forming reactions of electron deficient aryl iodides and phenylboronic acids. Furthermore, we demonstrate through exploration of a range of aryl iodides and boronic acids, using both competitive and non-competitive methodologies, that both the yields and the specific selectivity for the cross-coupled product depend critically on the steric properties of the aryl iodide reactant. As the confined reactions are seemingly independent of the properties of the boronic acid, we propose that nanoscale confinement in GNF affects only the oxidative addition step at the nanoreactor step-edge, with the reactivity of the afforded intermediate dependant on the steric congestion at the reaction locus. Thus, we show that the desirable properties of confinement can be modulated through considered selection of appropriate aryl iodide reactants.
Experimental
General experimental
All reagents were purchased from Sigma-Aldrich, UK and used without further purification. Carbon nanostructures were purchased from a range of commercial suppliers: SWNT (helix SWNT from Helix Material Solutions, made by arc discharge), MWNT (PD30L520 MWNT from NanoLab, made by chemical vapour deposition) and GNF (Pyrograf PR19 GNF from Applied Science Inc., made by chemical vapour deposition). All glassware was cleaned with a mixture of hydrochloric and nitric acid (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, ‘aqua regia’) and rinsed thoroughly with deionised water, cleaned with potassium hydroxide in isopropyl alcohol and finally rinsed with deionised water. 1H and 13C NMR spectra were obtained using a Bruker DPX-300 (300.13 and 75.48 MHz respectively) spectrometer at 298 K using CDCl3 as the solvent. Mass spectrometry was conducted on either a Bruker Apex IV (ESI) or a Waters Autospec (EI) in positive mode. Thermogravimetric analysis was performed using a TA Instruments SDT Q600 under a flow of air at a rate of 90 mL min−1 at a heating rate of 10 °C min−1 from room temperature to 1000 °C. Transmission electron microscopy (TEM) was performed using a JEOL 2100F TEM (field emission gun source, information limit <0.19 nm, 200 kV accelerating voltage) at room temperature. Statistical analysis of nanoparticle size (N > 50) and location was conducted using Gatan DigitalMicrograph software. Acquisition of tilt series was performed using a Gatan 916 tomography holder and a Gatan 646 Double tilt holder. This analysis involved tilting individual nanoreactor catalysts along the GNF growth axis and was used to determine the location of catalytic centres (inside or outside nanoreactors). Statistical relevance was ensured by analysing multiple nanoreactors, in different regions of the specimen and averaging across the entire sample. Energy dispersive X-ray analysis was performed using an Oxford Instruments INCA 560 X-ray microanalysis system. TEM samples were prepared by drop-drying methanolic solutions onto a copper grid mounted “lacey” carbon films.
:1 v/v, ‘aqua regia’) and rinsed thoroughly with deionised water, cleaned with potassium hydroxide in isopropyl alcohol and finally rinsed with deionised water. 1H and 13C NMR spectra were obtained using a Bruker DPX-300 (300.13 and 75.48 MHz respectively) spectrometer at 298 K using CDCl3 as the solvent. Mass spectrometry was conducted on either a Bruker Apex IV (ESI) or a Waters Autospec (EI) in positive mode. Thermogravimetric analysis was performed using a TA Instruments SDT Q600 under a flow of air at a rate of 90 mL min−1 at a heating rate of 10 °C min−1 from room temperature to 1000 °C. Transmission electron microscopy (TEM) was performed using a JEOL 2100F TEM (field emission gun source, information limit <0.19 nm, 200 kV accelerating voltage) at room temperature. Statistical analysis of nanoparticle size (N > 50) and location was conducted using Gatan DigitalMicrograph software. Acquisition of tilt series was performed using a Gatan 916 tomography holder and a Gatan 646 Double tilt holder. This analysis involved tilting individual nanoreactor catalysts along the GNF growth axis and was used to determine the location of catalytic centres (inside or outside nanoreactors). Statistical relevance was ensured by analysing multiple nanoreactors, in different regions of the specimen and averaging across the entire sample. Energy dispersive X-ray analysis was performed using an Oxford Instruments INCA 560 X-ray microanalysis system. TEM samples were prepared by drop-drying methanolic solutions onto a copper grid mounted “lacey” carbon films.
Preparation of PdNP@GNF nanoreactors
PdNP@GNF were prepared by modification of the method described by Zhi et al.42 A solution of potassium tetrachloropalladate (3.83 mg, 0.0117 mmol) in deionised water (1 mL) was added dropwise to graphitised nanofibres (25 mg, GNF, heated for 1 h at 450 °C in air). The combined dispersion was stirred at room temperature for 1 h and then dried at room temperature under vacuum for 15 h. The obtained solid was heated in a tube furnace under an atmosphere of hydrogen/argon (10/90) at 400 °C for 3 h and finally cooled at room temperature under an inert atmosphere of argon to yield a black solid. Additional catalytic nanoreactors and supported catalysts were prepared according to modified literature procedures and novel protocols (S1 and S2, ESI†).26,46–49The Suzuki–Miyaura cross-coupling reaction
The catalytic properties of nanoreactors and supported catalysts were determined using the Suzuki–Miyaura cross-coupling reaction (S3, ESI†).52 In a typical procedure, to the aryl iodide 1a–e (0.041–0.056 mmol, 1.0 eq.), the boronic acid 2a–e (0.053–0.075 mmol, 1.3 eq.), sodium acetate (0.9–17.2 mg, 0.094–0.13 mmol, 2.3 eq.) and the catalyst (0.2–4.7 mg, 2 mol%) in a two-necked round-bottomed flask under an inert atmosphere of argon was added via cannula a degassed solution of methanol (5 mL) and the resulting suspension stirred at 70 °C for 24 h. After cooling to room temperature, the dispersion was filtered through a PTFE membrane filter (pore diameter 0.2 μm) and concentrated under vacuum. The products were identified by 1H NMR spectroscopy of the crude mixture or isolated by flash column chromatography on a silica gel (ccsg). Competitive Suzuki–Miyaura reactions were performed in an analogous fashion to the method described above (S3, ESI†).Results and discussion
Graphitised nanofibres are an ideal material to compare the effects of catalyst support and confinement on the properties of the Suzuki–Miyaura cross-coupling reaction given the significant difference between their external and internal surfaces. This should permit discrete control of the location of catalyst centres through careful consideration of catalyst assembly conditions. Furthermore, the cost of GNF is relatively low and the presence of nanofibre synthesis catalyst, which has the potential to adversely affect the desired properties of the nanocatalyst, is negligible. In our preliminary experiments, a range of novel catalysts comprising palladium nanoparticles confined for the first time within the internal channel of GNF were prepared based on adaptation to literature procedures for the encapsulation of palladium in CNT26,46–49 and mesoporous silica.42 The loading of palladium precursors was fixed across all samples and the structural properties of the obtained composites were evaluated by extensive transmission electron microscopy imaging (Fig. 1 and 3, Table 1 and Fig. S1 and S2, ESI†) and thermogravimetric analysis (Table 1 and Fig. S3, ESI†). | ||
| Fig. 1 TEM images of the catalytic carbon nanoreactors: (a) PdNP@GNF possessing small PdNP (dNP = 7.0 ± 2.2 nm) which nucleate and grow at the graphitic step-edges of GNF nanoreactors; (b) PdNP@GNF-I comprising a bimodal distribution of small pseudo-spherical PdNP (dNP = 15.3 ± 8.9 nm) and larger faceted nanoparticles which are mostly located on the nanofibre exterior; (c) PdNP@GNF-II possessing PdNP (dNP = 9.4 ± 6.4 nm) mainly located on the outer surface of GNF, owing to poor impregnation of the GNF suspension with the palladium precursor solution; (d) PdNP@GNF-III assembled using gas-phase impregnation of GNF with the palladium precursor, leading to the formation of PdNP (dNP = 12.9 ± 3.9 nm) poorly inserted inside GNF and covered with carbon shells and (e) PdNP@GNF-IV characterised by ultra-small PdNP (dNP = 1.1 ± 0.3 nm) pre-formed and selectively encapsulated inside GNF by using scCO2 as transport and process medium. Scale bars are (a) 50, (b–d) 100 and (e) 20 nm. | ||
| Entry | Catalyst | TEM analysis | TGA analysis | ||
|---|---|---|---|---|---|
| d NP /nm | PdNP locationb | T oxid /°C | Pd900c/% | ||
| a d NP refers to the mean nanoparticle diameter as determined by TEM. b PdNP location was estimated by tilt analysis (Fig. S2, ESI). c T oxid and Pd900 refer to the temperature of primary oxidation in air and the percentage of residual palladium metal at 900 °C respectively as determined by TGA. | |||||
| 1 | PdNP@GNF | 7.0 ± 2.2 | Mostly in (>80%) | 705 | 7.8 |
| 2 | PdNP@GNF-I | 15.3 ± 8.9 | Mostly out | 711 | 9.5 |
| 3 | PdNP@GNF-II | 9.4 ± 6.4 | Mostly out | 691 | 6.2 |
| 4 | PdNP@GNF-III | 12.9 ± 3.9 | Mostly out | 737 | 8.2 |
| 5 | PdNP@GNF-IV | 1.1 ± 0.3 | Mostly in (>90%) | 711 | 4.1 |
PdNP@GNF, PdNP@GNF-I and PdNP@GNF-II were prepared by liquid-phase impregnation of GNF, either as a solid or in aqueous suspension, with an aqueous solution of potassium tetrachloropalladate, followed by removal of the solvent and treatment with molecular hydrogen to form ligand-free, and therefore highly reactant accessible, metal nanoparticles.42,46,47 PdNP@GNF-III was prepared by gas-phase impregnation of GNF with palladium bis(acetylacetonate) and subsequent hydrogen-mediated reduction48 and PdNP@GNF-IV by the encapsulation of pre-formed dodecanethiolate-stabilised palladium nanoparticles49 using scCO2 as a transport and processing medium.26
Comprehensive transmission electron microscopy investigation of the structure of the composite materials reveals that PdNP@GNF-I, -II and -III (Fig. 1(b–d) and S1(a–c), ESI†) were not suitable to study the confined Suzuki–Miyaura reactions as the majority of the palladium catalytic centres appear on the external surfaces of nanofibres, with the catalytically active surface passivated by graphitic carbon shells in some cases (Fig. S1(c), ESI†). Conversely, nanoparticle catalysts in PdNP@GNF (Fig. 1(a), 3(a, b) and S2, ESI†) and PdNP@GNF-IV (Fig. 1(e) and S1(d), ESI†) are nearly exclusively located at the internal corrugations within GNF and hence represent more attractive materials to explore the properties of confined cross-coupling reactions.
The catalytic performance of palladium nanoparticle–graphitised nanofibre nanoreactors was appraised using the reaction of 1-iodo-4-nitrobenzene 1a and phenylboronic acid 2a (Scheme 1 and Table 2).
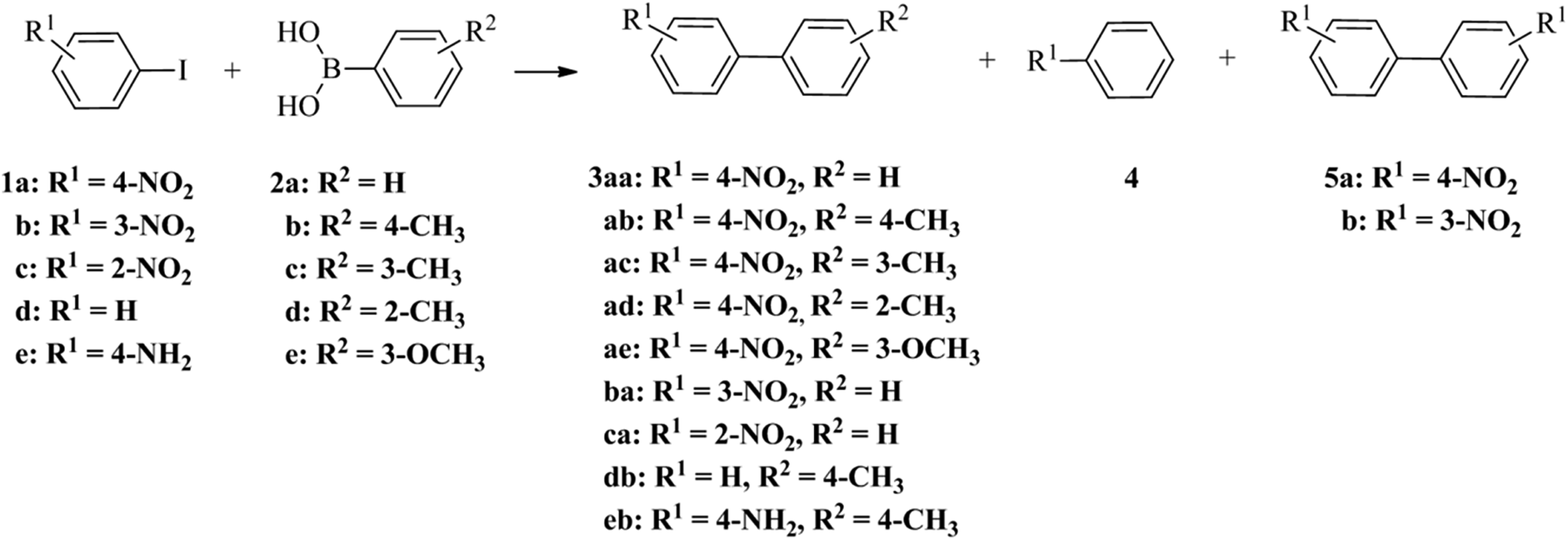 | ||
| Scheme 1 The palladium catalysed reactions of aryl iodides 1 and phenylboronic acids 2 leading to the products of Suzuki–Miyaura cross-coupling 3, dehalogenation 4 and Ullmann coupling 5. | ||
| Entry | Catalyst | t/h | Conversionb/% | Selectivity for 3aa (4:5a)b/% |
|---|---|---|---|---|
| a Standard conditions: 1-iodo-4-nitrobenzene 1a (0.056 mmol, 1.0 eq.), phenylboronic acid 2a (0.075 mmol, 1.3 eq.), sodium acetate (0.13 mmol, 2.3 eq.), catalyst (2 mol%), methanol (5 mL), 70 °C, 24 h. b Conversion and selectivity for 4-nitrobiphenyl 3aa, nitrobenzene 4 and 4,4′-dinitrobiphenyl 5a, the products of cross-coupling, dehalogenation and Ullmann coupling respectively, were determined by 1H NMR spectroscopy of the crude mixture. | ||||
| 1 | PdNP@GNF | 24 | 83 | 87 (10:3) |
| 2 | PdNP@GNF-I | 24 | 98 | 95 (0:5) |
| 3 | PdNP@GNF-II | 24 | 82 | 96 (0:4) |
| 4 | PdNP@GNF-III | 24 | 36 | 93 (2:5) |
| 5 | PdNP@GNF-IV | 144 | 40 | 0 (100:0) |
Although the assembly of nanoreactors by insertion of pre-formed nanoparticles is generally preferred to methodologies involving direct formation of nanoparticles in nanoreactors, due to the retention of structural properties of the pre-formed catalysts regardless of their explicit location (inside or outside nanoreactors), the reaction catalysed by PdNP@GNF-IV was sluggish, resulting in low conversion of starting materials at extended reaction times and affording only the by-product of dehalogenation 4 (Table 2, entry 5). The significant reduction in catalytic performance of PdNP@GNF-IV, likely associated with the poor accessibility of reactants to the catalyst due to a combination of deep penetration of small nanoparticles at the step-edge reaction locus and the presence of a sterically-congesting nanoparticle stabilisation agent, means that these catalytic materials were unsuitable for further use. The reactions catalysed by the ligand-free catalysts PdNP@GNF-I and -II resulted in high conversion and selectivity for the cross-coupled product 3aa (Table 2, entries 2 and 3, respectively) but do not allow investigation of the effects of nanoscale confinement due to the location of the catalytic centres almost exclusively on the exterior surfaces, whereas the passivation of the catalyst by carbon shells resulted in the poor catalytic properties of PdNP@GNF-III (Table 2, entry 4). Conversely, the reaction catalysed by PdNP@GNF proceeded with good conversion and selectivity for the elected cross-coupled product (Table 2, entry 1), demonstrating catalytic properties comparable to more traditional molecular palladium catalysts, e.g. PdCl2(PPh3)2.50 Furthermore, and most significantly, recycling of the catalyst in the reaction of 1-iodo-4-cyanobenzene and phenylboronic acid indicated that confinement of nanoparticles at the nanofibre step-edges preserves the selectivity for the cross-coupled product across catalytic cycles and, despite a moderate drop in activity after the initial reaction, retains good activity cycle-to-cycle over at least five catalytic cycles (Fig. 2 and S3.2, ESI†). Thus, PdNP@GNF represents a superior catalytic material relative to classical molecular catalysts which cannot be recycled.
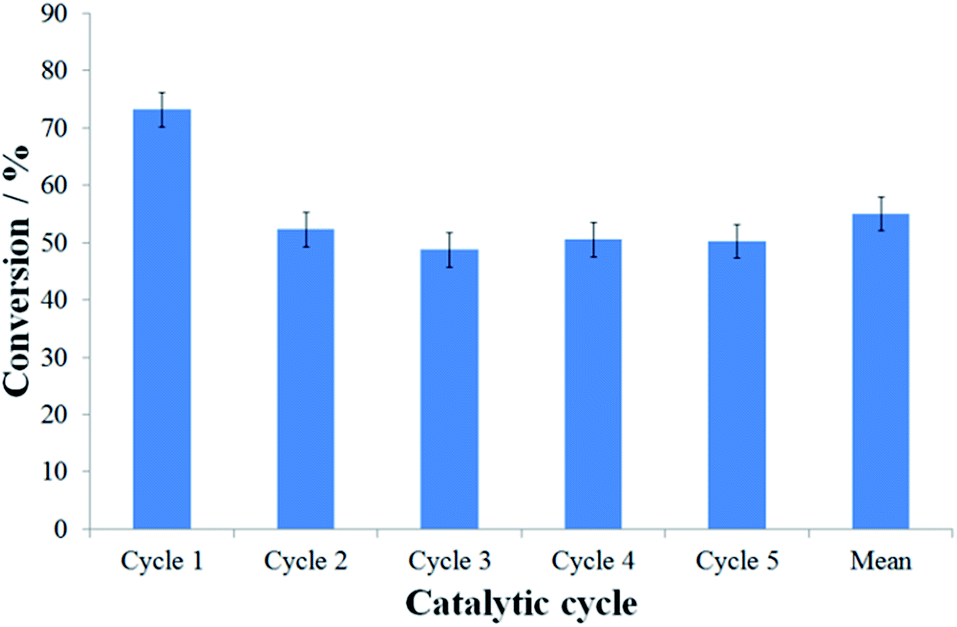 | ||
| Fig. 2 Evaluation of the conversion in the Suzuki–Miyaura cross-coupling of 1-iodo-4-cyanobenzene and phenylboronic acid after 16 h using PdNP@GNF nanoreactors over five catalytic cycles. The selectivity for the cross-coupled product 4-cyanobiphenyl was 100% across all catalytic cycles. | ||
HR-TEM imaging of the PdNP@GNF structures after one catalytic cycle (Fig. S4, ESI†) illustrates that the initial small PdNP (dNP = 7.0 ± 2.2 nm) undergo a process of growth into larger nanoparticles (dNP = 21.1 ± 9.8 nm), modulated by Ostwald ripening under the so-called “dissolution-precipitation equilibrium” typically observed in nanoparticle catalysis. This explains the slight decrease in activity after the first cycle, but critically the nanoparticle catalysts remain anchored to the step-edges within GNF and therefore are available for subsequent reactions. Indeed, nanoparticle size (dNP = 21.8 ± 14.8 nm) does not seemingly change after five catalytic cycles (Fig. 3(c, d) and S4, ESI†), highlighting the high stability of the catalyst, imparted by confinement at nanoreactor step-edges, which is reflected in the consistent activities observed after cycle one.
 | ||
| Fig. 3 TEM analysis of PdNP@GNF catalytic nanoreactors (a and b) before catalysis and (c and d) after five catalytic cycles showing PdNP growth from 7.0 ± 2.2 to 21.8 ± 14.8 nm. The corrugated interior of GNF facilitates favourable van der Waals interactions with encapsulated nanoparticles, modulating the controlled growth of nanoparticle catalytic centres and restricting the loss of precious metal from the internal channel of the nanoreactor under the conditions of the Suzuki–Miyaura reaction. Inset: schematic representation of the location of palladium nanoparticles at the internal step-edges of GNF. The proposed reaction locus is denoted by a red circle at the apex of the two graphitic surfaces of the nanofibre step-edge and the catalytic centre. Scale bars are (a and d) 50 and (b and c) 10 nm. | ||
As the PdNP@GNF composite exhibited effective control of catalyst positioning, good stability and promising catalytic properties in preliminary cross-coupling reactions, we decided to fabricate the corresponding material with nanoparticles deposited on the atomically-smooth exterior surface of GNF (PdNP/GNF) in order to facilitate investigation of the effect of nanoscale confinement on the Suzuki–Miyaura reaction (Fig. S5(d), ESI†). Furthermore, we explored the versatility of the preparation protocol of PdNP@GNF for the encapsulation of catalyst nanoparticles in narrower carbon nanostructures, such as single- and multi-walled carbon nanotubes (SWNT and MWNT, respectively). Confinement of palladium nanoparticles failed within SWNT likely owing to the small internal diameter of these narrow nanotubes and the consequent high resistance to the transport of precursor molecules into the internal channel (Fig. S5(c), ESI†). Indeed, palladium nanoparticles were observed within MWNT but, the catalytic material almost completely blocked the inner channel of the nanotube, thus inhibiting the penetration of molecular reactants (Fig. S5(b), ESI†). Hence, only GNF-based nanoscale catalysts were further explored.
To probe the influence of nanoscale confinement in GNF on the properties of the Suzuki–Miyaura cross-coupling reaction, we conducted a comprehensive study where the steric and electronic properties of both the aryl iodide and boronic acid coupling partners were systematically varied and the yields and distribution of products compared for reactions catalysed by PdNP@GNF nanoreactors and PdNP/GNF supported catalysts (Scheme 1 and Table 3).
| Entry | Catalyst | R1 | R2 | Conversionb/% | Selectivity for 3 (4:5a–b)b/% |
|||
|---|---|---|---|---|---|---|---|---|
| a Standard conditions: aryl iodides 1a–e (0.041–0.056 mmol, 1.0 eq.), boronic acids 2a–e (0.053–0.075 mmol, 1.3 eq.), sodium acetate (0.094–0.13 mmol, 2.3 eq.), catalyst (2 mol%), methanol (5 mL), 70 °C, 24 h. b Conversion and selectivity for biphenyls 3aa–eb, nitrobenzene 4 and 4,4′- and 3,3′-dinitrobiphenyls 5a and 5b, respectively, the products of cross-coupling, dehalogenation and Ullmann coupling respectively were determined by 1H NMR spectroscopy of the crude mixture. | ||||||||
| 1 | PdNP@GNF | 1a | 4-NO2 | 2a | H | 83 | 3aa | 87 (10:3) |
| 2 | PdNP/GNF | 1a | 4-NO2 | 2a | H | 78 | 3aa | 80 (17:3) |
| 3 | PdNP@GNF | 1a | 4-NO2 | 2b | 4-Me | 77 | 3ab | 94 (3:3) |
| 4 | PdNP/GNF | 1a | 4-NO2 | 2b | 4-Me | 65 | 3ab | 92 (4:4) |
| 5 | PdNP@GNF | 1a | 4-NO2 | 2c | 3-Me | 74 | 3ac | 97 (1:2) |
| 6 | PdNP/GNF | 1a | 4-NO2 | 2c | 3-Me | 74 | 3ac | 97 (1:2) |
| 7 | PdNP@GNF | 1a | 4-NO2 | 2d | 2-Me | 58 | 3ad | 68 (32:0) |
| 8 | PdNP/GNF | 1a | 4-NO2 | 2d | 2-Me | 58 | 3ad | 67 (33:0) |
| 9 | PdNP@GNF | 1a | 4-NO2 | 2e | 3-OMe | 83 | 3ae | 96 (2:2) |
| 10 | PdNP/GNF | 1a | 4-NO2 | 2e | 3-OMe | 81 | 3ae | 95 (3:2) |
| 11 | PdNP@GNF | 1b | 3-NO2 | 2a | H | 99 | 3ba | 71 (28:1) |
| 12 | PdNP/GNF | 1b | 3-NO2 | 2a | H | 98 | 3ba | 79 (19:2) |
| 13 | PdNP@GNF | 1c | 2-NO2 | 2a | H | 80 | 3ca | 64 (36:0) |
| 14 | PdNP/GNF | 1c | 2-NO2 | 2a | H | 87 | 3ca | 99 (1:0) |
| 15 | PdNP@GNF | 1d | H | 2b | 4-Me | 42 | 3db | 100 (0:0) |
| 16 | PdNP/GNF | 1d | H | 2b | 4-Me | 44 | 3db | 100 (0:0) |
| 17 | PdNP@GNF | 1e | 4-NH2 | 2b | 4-Me | 23 | 3eb | 100 (0:0) |
| 18 | PdNP/GNF | 1e | 4-NH2 | 2b | 4-Me | 24 | 3eb | 100 (0:0) |
The effect of substituent position on the aryl iodide was studied by performing the reactions of 1-iodo-4-, 1-iodo-3- or 1-iodo-2-nitrobenzene 1a–c with phenylboronic acid 2a (Table 3, entries 1 vs. 2, 11 vs. 12 and 13 vs. 14). Subtle enhancements in the conversion and selectivity for the cross-coupled product in the reaction of the non-sterically hindered 1-iodo-4-nitrobenzene 1a and 2a were observed for reactions catalysed by PdNP@GNF. Slight improvements in conversion were seen for the confined reaction of 1-iodo-3-nitrobenzene 1b and 2a. However, the reaction of the sterically congested 1-iodo-2-nitrobenzene 1c with 2a was seemingly more sensitive to catalyst location, with a subtle reduction in conversion and a notable decrease in selectivity for the corresponding cross-coupled product 3ca observed for the confined reaction. Interestingly, no effect of confinement was observed when comparing the catalyst-confined and catalyst-supported reactions of 1-iodo-4-nitrobenzene 1a with 4-, 3- and 2-tolylboronic acids 2b–d (Table 3, entries 3 vs. 4, 5 vs. 6 and 7 vs. 8). It is important to note that, although the reaction of bulky 2-tolylboronic acid 2d was less selective and proceeded more slowly compared to the cross-couplings of 4- and 3-tolylboronic acids 2b and c, presumably due to increased steric hindrance of the incoming boronic acid to the oxidative addition intermediate, this behaviour was independent of the reaction locus (i.e. inside or outside nanoreactors) whereas for the sterically hindered 1-iodo-2-nitrobenzene 1c the PdNP/GNF supported catalyst was a superior catalyst than the PdNP@GNF confined catalyst (Table 3, entry 13 vs. 14).
In order to investigate whether nanoscale confinement affected solely the reaction pathway of sterically hindered aryl iodides, we also explored the effect of the nature of the substituent of both the aryl iodide and the boronic acid and thus the potential influence of electronic effects on the cross-coupling reactions. Comparison of the catalysed reactions of 1-iodo-4-nitrobenzene 1a, iodobenzene 1d and 4-iodoaniline 1e with 4-tolylboronic acid 2b (Table 3, entries 1 vs. 2, 15 vs. 16 and 17 vs. 18) evidences that confinement does not change the pathways of these reactions, with activities following the known reactivity profiles (i.e. NO2 > H > NH2) and selectivities according to the selectivity-reactivity principle for cross-coupling reactions51 using both PdNP@GNF and PdNP/GNF. Further evidence of the scarce influence of both substituent position and electronic effect on the boronic acid was obtained by performing the reaction of 1a with 3-methoxybenzene 2e (Table 3, entry 9 vs. 10). We have observed that irrespective of the boronic acid employed, neither the electronic or steric features led to any significant changes in the properties of the reaction induced by confinement. It is interesting to note that Ullmann homo-coupling products 5a–b were also detected in our exploration of catalyst properties; however, these products are generally only present in minor quantities, particularly for the reactions of activated and non-bulky aryl iodides 1a–b, and their formation critically is seemingly unaffected by nanoscale confinement in nanoreactors.
In order to rationalise the behaviour observed in our study, it is important to carefully consider the mechanism of the palladium nanoparticle-mediated Suzuki–Miyaura cross-coupling reaction. Whilst the specific nature of the active species and mode of operation remains somewhat controversial in the scientific literature,52 with leading researchers providing contradictory evidence to support homogeneous53–55 and heterogeneous56–58 mechanistic pathways, it is becoming increasingly accepted that nanoparticle-catalysed cross-coupling reactions follow a quasi-homogeneous pathway,59 wherein the catalytic cycle is initiated by the oxidative addition of low coordinate surface Pd atoms to the aryl halide reactant. This results in leaching of soluble palladium species into solution, such as [ArPdX] and [ArPdX2]− (where Ar = aryl, X = halide), which undergo further transformations, the rate and nature of which are influenced by the experimental conditions, the concentrations of specific components of the reaction mixture and the properties of the catalyst.
As the catalytic centre in PdNP@GNF is located at the internal step-edge, it follows logically that the first step of the catalytic cycle, the oxidative addition, must take place at the nanoparticle–nanofibre interface. Here, π–π interactions between the incoming reactant and the graphitic surfaces of the nanoreactor step-edge are prevalent and facilitate high local concentrations and pre-organisation of the substrate prior to oxidative addition.24–27 Our measurements provide compelling evidence of the prevailing role of the oxidative addition step in the nanoparticle-mediated Suzuki–Miyaura cross-coupling reaction. We propose that the observed increase in activity in nanoreactors for non-sterically demanding aryl iodide substrates (Table 3, entries 1 vs. 2 and 3 vs. 4) is associated with enhanced concentration of both aryl iodide reactants and soluble palladium species at the nanofibre step-edge; conversely, the conversion for reactions performed outside nanofibres is higher for more sterically-congested structural analogues (Table 3, entry 13 vs. 14) due to unrestrained aryl concentration outside the nanofibres. More interestingly, the selectivity for the products of cross-coupling 3ba and 3ca and dehalogenation 4 also appears to be dependent on the steric properties of the aryl iodide reactants, with more congested aryl iodides promoting the dehalogenation process, particularly for the cross-coupling of bulky 1-iodo-2-nitrobenzene 1c (Table 3, entries 11 vs. 12 and 13 vs. 14). This unexpected behaviour is attributed to significant molecular crowding for sterically congested aryl iodides inside nanoreactors. Here, the leached oxidative addition intermediate remains trapped in the restricted volume of the step-edge and therefore its coordination to the incoming boronic acid becomes partially impeded. Consequently, rapid diffusion of methanol to the step-edge, due to its reduced molecular size becomes more favourable, promoting the formation of the product of dehalogenation (Scheme 2).60–64
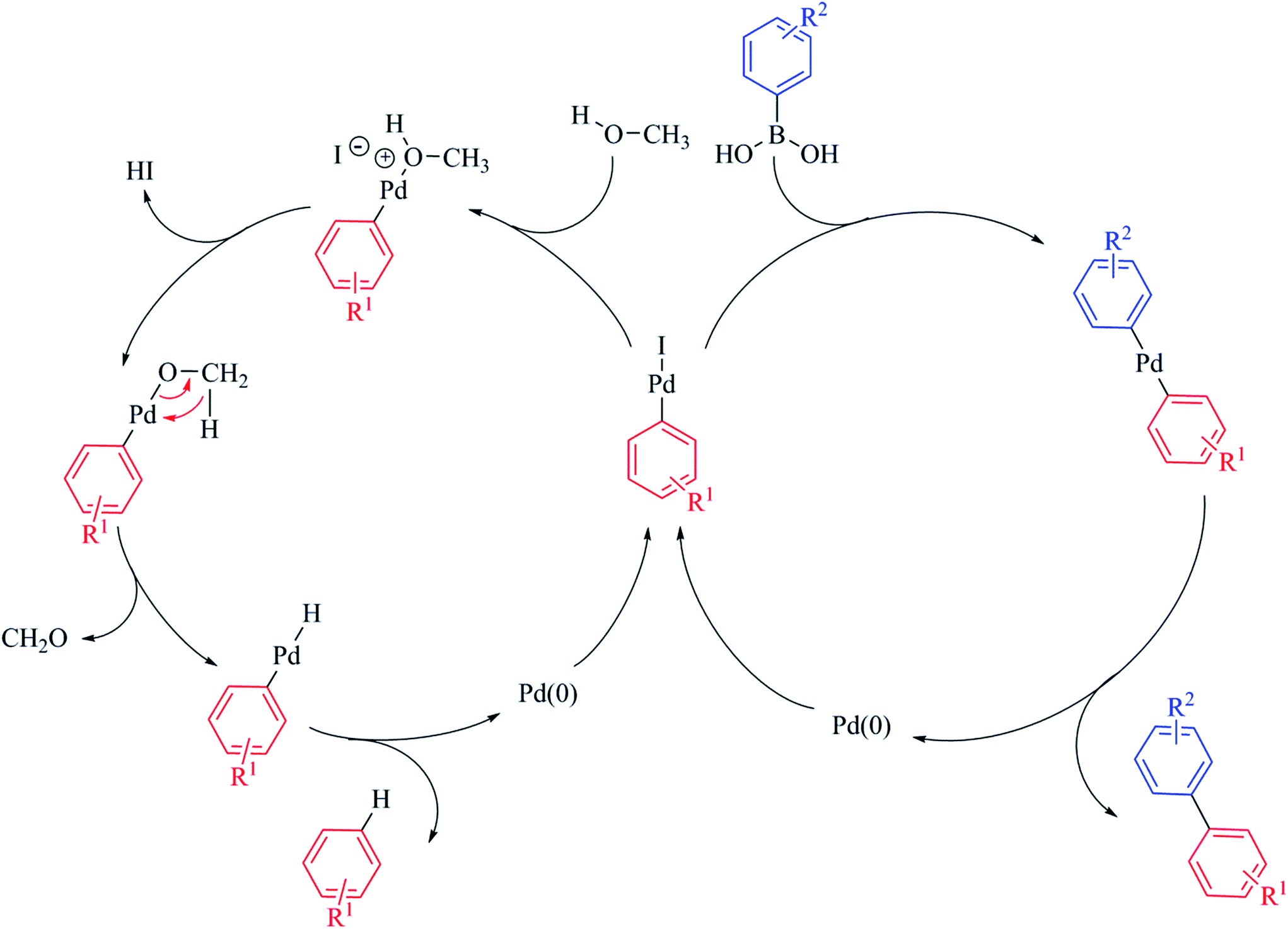 | ||
| Scheme 2 The fate of the intermediate following oxidative addition yields the products of cross-coupling or dehalogenation depending on the competitive reactions with boronic acid or methanol respectively. The dehalogenation reaction is initiated by displacement of iodide from the oxidative addition intermediate by methanol. Subsequent to deprotonation, β-hydride elimination affords the [ArPdH] intermediate from which the product of dehalogenation ArH is obtained.64 It is important to note that both mechanistic pathways result in the elimination of Pd(0), which is retained within the nanoreactor channel and re-deposits onto existing nanoparticles confined at the internal step-edges. Thus, the observed Ostwald ripening of nanoparticles subsequent to catalysis provides further evidence supporting the quasi-homogeneous nature of nanoparticle-catalysed reactions. | ||
Indeed, for highly congested step-edge environments, such as those using PdNP@GNF-IV, only the product of dehalogenation is observed (Table 2, entry 5). This provides further evidence for the importance of the step-edge environment in modulating the pathways of chemical reactions, with the rate of confined reactions determined by oxidative addition and the selectivity critically dependent on the steric properties of the aryl halide. This implies that, subsequent to oxidative addition, further reactions do not occur within the step-edge, but rather within the internal cavity of the nanoreactor. In this larger reaction volume, the pathway of the reaction will therefore be unaffected by the steric properties of the boronic acid and hence the effects of confinement on activity and selectivity will not be observed.
To further probe this phenomenon, we performed a series of competitive Suzuki–Miyaura reactions, where either two aromatic boronic acids compete for a common aryl iodide or two aryl iodides for a common boronic acid (Scheme 3 and Table 4).
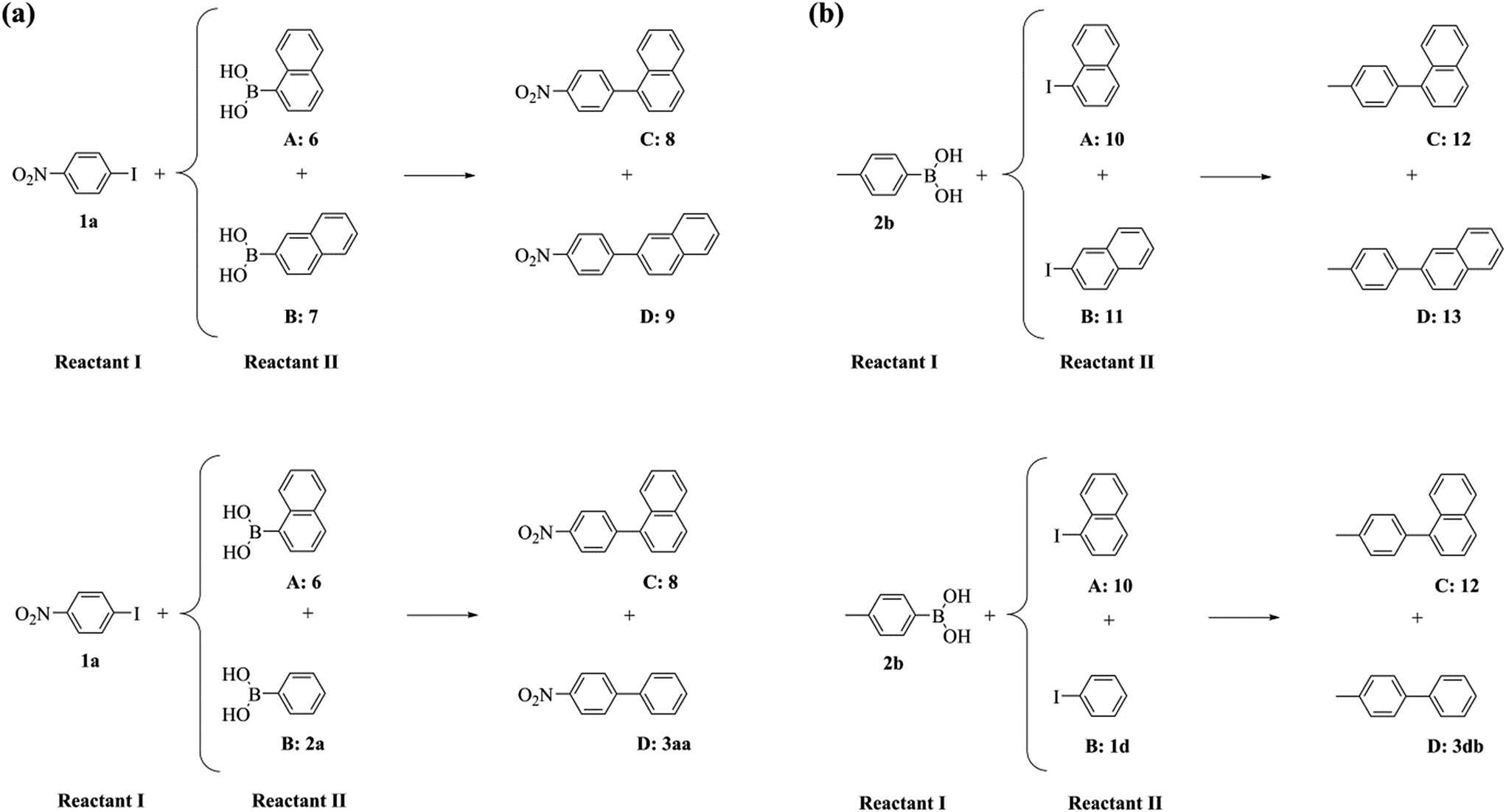 | ||
| Scheme 3 The competitive Suzuki–Miyaura reactions of (a) two aromatic boronic acids with a common aryl iodide and (b) two aryl iodides with a common aromatic boronic acid. | ||
| Entry | Catalyst | Reactant I | Reactant II | Conversionb/% | Selectivity for cross-coupled productsb/% (C:D) |
|
|---|---|---|---|---|---|---|
| A | B | |||||
| a Standard conditions: 1-iodo-4-nitrobenzene 1a, 1-naphthalene boronic acid 6, 2-naphthalene boronic acid 7, phenylboronic acid 2a, 4-tolylboronic acid 2b, 1-iodonaphthalene 10, 2-iodonaphthalene 11 and iodobenzene 1d (0.065 mmol, 1 eq.), sodium acetate (0.15 mmol, 2.3 eq.), catalyst (2 mol%), methanol (5 mL), 70 °C, 24 h. b Determined by 1H NMR spectroscopy of the crude mixture. | ||||||
| 1 | PdNP@GNF | 1a | 6 | 7 | 63 | 28:72 (8:9) |
| 2 | PdNP/GNF | 1a | 6 | 7 | 63 | 28:72 (8:9) |
| 3 | PdNP@GNF | 1a | 6 | 2a | 67 | 63:37 (8:3aa) |
| 4 | PdNP/GNF | 1a | 6 | 2a | 68 | 64:36 (8:3aa) |
| 5 | PdNP@GNF | 2b | 10 | 11 | 94 | 15:85 (12:13) |
| 6 | PdNP/GNF | 2b | 10 | 11 | 89 | 42:58 (12:13) |
| 7 | PdNP@GNF | 2b | 10 | 1d | 52 | 100:0 (12:3db) |
| 8 | PdNP/GNF | 2b | 10 | 1d | 53 | 80:20 (12:3db) |
The reaction of 1-iodo-4-nitrobenzene 1a and either 1- or 2-naphthalene boronic acids, 6 and 7 respectively, exhibited the same conversion of 1a and selectivity ratio between the afforded cross-coupled products 8 and 9 using both the PdNP catalytic nanoreactor and the supported catalyst (Table 4, entry 1 vs. 2). Furthermore, the cross-coupling of 1a with either 1-naphthalene or phenylboronic acids, 6 or 2a respectively, was similarly unaffected by confinement (Table 4, entry 3 vs. 4). These results confirm that confinement effects are independent of the nature of the boronic acid. In contrast, when two aryl iodides compete for a common boronic acid, significant effects induced by confinement were observed. Whilst the reactions of 4-tolylboronic acid 2b and a mixture of 1- and 2-iodonaphthalenes, 10 and 11 respectively, showed comparable conversion of 2b irrespective of the catalyst location, confinement of the catalyst resulted in a 2.8-fold decrease in selectivity towards the formation of the cross-coupled product with bulky 1-iodonaphthalene 10 compared to the reaction catalysed by PdNP/GNF (Table 4, entry 5 vs. 6). In addition, comparison of the properties of the confined and supported reactions of 4-tolylboronic acid 2b with both 1-iodonaphthalene 10 and iodobenzene 1d (Table 4, entry 7 vs. 8), indicated that a higher selectivity for the cross-coupling of bulky 1-iodonaphthalene 10 was afforded. Clearly, the interactions of the naphthalene system with the graphitic surfaces became more important and maximal at the step-edges where the high local concentration of 1-iodonaphthalene 10 promoted a 6.7-fold increase in the selectivity towards the formation of the cross-coupled product 12 within the catalytic nanoreactor (Table 4, entry 5 vs. 7) in contrast to the 1.9-fold increase obtained for the reaction catalysed by supported PdNP (Table 4, entry 6 vs. 8). These results validate the hypothesis that the nature of the aryl iodides in confined reactions is crucial only if the oxidative addition step of the Suzuki–Miyaura reaction occurs at the GNF step-edge, where both the concentration and steric properties of the halide coupling partner can be harnessed to modify the pathway of the reaction.
Conclusions
We have investigated different preparation protocols for the optimisation of PdNP@GNF nanoreactors. We found that the aqueous impregnation of a palladium salt in GNF, followed by gas-phase reduction, enabled near selective confinement of palladium nanoparticles within the interior of graphitised nanofibres and the resultant catalytic nanoreactor exhibited attractive catalytic properties in the Suzuki–Miyaura cross-coupling reaction. A slight modification of this procedure allowed us to prepare relevant catalysts with nanoparticles deposited on the exterior of the nanoreactors in order to properly investigate the effect of confinement in cross-coupling reactions. Through systematic comparison of the yields and distributions of products of reactions using a series of substituted aryl iodides and boronic acids, we have demonstrated that the activity and selectivity of the confined reactions are sensitive to the steric congestion of the aryl iodides, whereas no influence of confinement was observed for the reaction of bulky boronic acids. The competitive Suzuki–Miyaura reactions, where two reagents compete for a common coupling agent, indicated that the confinement effects are only prevalent for two aryl iodides competing for one boronic acid and reside in the mechanism of confined cross-couplings. We have shown that the oxidative addition step of the catalytic cycle occurs at the GNF step-edge, where the local concentration and organisation of the aryl iodide reactant are critically influenced by nanoscale confinement, with the reactions of non-sterically demanding substrates promoted in this unusual reaction environment. Controlling the properties of organic transformations utilising the effects of catalyst confinement in graphitised nanofibres is becoming an increasingly versatile tool for synthetic chemistry, particularly as the unique step-edge reaction site has the potential to enhance both the activity and selectivity of the catalyst and promotes the recycle and reuse of the material multiples times with retention of elected catalyst properties.Acknowledgements
The authors would like to thank the European Research Council (ERC), the Engineering and Physical Sciences Research Council (EPSRC), the Italian Ministry of Education, University and Research (MIUR, PRIN 2010–2011 – prot. 2010N3T9M4) for supporting this research. BC is grateful for financial support from “Italian-French University” (Vinci 2011), University of Chieti-Pescara, “Centre National de la Research Scientifique (CNRS)” and University of Reims. The authors thank the Nottingham Nanotechnology and Nanoscience Centre (NNNC) for access to transmission electron microscopy facilities.Notes and references
- A. N. Khlobystov, ACS Nano, 2011, 5, 9306 CrossRef CAS PubMed.
- P. Serp and E. Castillejos, ChemCatChem, 2010, 2, 41 CrossRef CAS.
- X. Pan and X. Bao, Acc. Chem. Res., 2011, 44, 553 CrossRef CAS PubMed.
- D. A. Britz and A. N. Khlobystov, Chem. Soc. Rev., 2006, 35, 637 RSC.
- A. Zecchina, S. Bordiga and E. Groppo, Selective Nanocatalysts and Nanoscience, Wiley-VCH, Weinheim, 2011 Search PubMed.
- U. H. L. Brinker and J.-L. Mieusset, Molecular Encapsulation: Organic Reactions in Constrained Systems, Wiley, New York, 2010 Search PubMed.
- J. Lee, J. C. Park, J. U. Bang and H. Song, Chem. Mater., 2008, 20, 5839 CrossRef CAS.
- S. Shi, M. Wang, C. Chen, J. Gao, H. Ma, J. Ma and J. Xu, Chem. Commun., 2013, 49, 9591 RSC.
- S. Guernelli, R. Zappacosta, G. Siani, D. Spinelli and A. Fontana, J. Mol. Catal. A: Chem., 2014, 383–384, 114 CrossRef CAS PubMed.
- P. Marracino, A. Amadei, F. Apollonio, G. d'Inzeo, M. Liberti, A. Di Crescenzo, A. Fontana, R. Zappacosta and M. Aschi, J. Phys. Chem. B, 2011, 115, 8102 CrossRef CAS PubMed.
- M. Salvati-Niasari and A. Sobhani, J. Mol. Catal. A: Chem., 2008, 285, 58 CrossRef PubMed.
- K. Balasubramanian and M. Burgharo, Small, 2005, 1, 180 CrossRef CAS PubMed.
- R. S. Ruoff and D. C. Lorents, Carbon, 1995, 33, 925 CrossRef CAS.
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Carbon, 2006, 44, 1624 CrossRef CAS PubMed.
- M. Peer, A. Qajar, B.-P. M. Holbrook, R. Rajagopalan and H. C. Foley, Carbon, 2013, 57, 485 CrossRef CAS PubMed.
- B. W. Smith, M. Monthioux and D. E. Luzzi, Nature, 1998, 396, 323 CrossRef CAS PubMed.
- S. Ittisanronnachai, H. Orikasa, N. Inokuma, Y. Uozu and T. Kyotani, Carbon, 2008, 46, 1361 CrossRef CAS PubMed.
- K. Schulte, J. Swarbrick, N. Smith, F. Bondino, E. Magnano and A. N. Khlobystov, Adv. Mater., 2007, 19, 3312 CrossRef CAS.
- R. Marega and D. Bonifazi, New J. Chem., 2014, 38, 22 RSC.
- A. N. Khlobystov, D. A. Britz and G. A. D. Briggs, Acc. Chem. Res., 2005, 38, 901 CrossRef CAS PubMed.
- S. A. Miners, G. A. Rance and A. N. Khlobystov, Chem. Commun., 2013, 49, 5586 RSC.
- E. Castillejos, P.-J. Debouttiere, L. Roiban, A. Solhy, V. Martinez, Y. Kihn, O. Ersen, K. Philippot, B. Chaudret and P. Serp, Angew. Chem., Int. Ed., 2009, 48, 2529 CrossRef CAS PubMed.
- E. Castillejos, R. Chico, R. Bacsa, S. Coco, P. Espinet, M. Perez-Cadenas, A. Guerrero-Ruiz, I. Rodriguez-Ramos and P. Serp, Eur. J. Inorg. Chem., 2010, 5096 CrossRef CAS.
- A. La Torre, M. C. Gimenez-Lopez, M. Fay, G. A. Rance, W. A. Solomonsz, T. W. Chamberlain, P. D. Brown and A. N. Khlobystov, ACS Nano, 2012, 6, 2000 CrossRef CAS PubMed.
- G. A. Rance, W. A. Solomonsz and A. N. Khlobystov, Chem. Commun., 2013, 49, 1067 RSC.
- W. A. Solomonsz, G. A. Rance, M. Suyetin, A. La Torre, E. Bichoutskaia and A. N. Khlobystov, Chem.–Eur. J., 2012, 18, 13180 CrossRef CAS PubMed.
- W. A. Solomonsz, G. A. Rance, B. J. Harris and A. N. Khlobystov, Nanoscale, 2013, 5, 12200 RSC.
- A. La Torre, M. W. Fay, G. A. Rance, M. C. Gimenez-Lopez, W. A. Solomonsz, P. D. Brown and A. N. Khlobystov, Small, 2012, 8, 1222 CrossRef CAS PubMed.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 15, 2419 Search PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC.
- A. O. King and N. Yasuda, Top. Organomet. Chem., 2004, 6, 205 CAS.
- N. Yasuda, J. Organomet. Chem., 2002, 653, 279 CrossRef CAS.
- R. W. Friesen, C. Brideau, C. C. Chan, S. Charleson, D. Deschênes, D. Dubé, D. Ethier, R. Fortin, J. Y. Gauthier, Y. Girard, R. Gordon, G. M. Greig, D. Riendeau, C. Savoie, Z. Wang, E. Wong, D. Visco, L. J. Xu and R. N. Young, Bioorg. Med. Chem. Lett., 1998, 8, 2777 CrossRef CAS.
- T. J. Colacot, Platinum Met. Rev., 2011, 55, 84 CrossRef.
- M. Garcia-Melchor, A. A. C. Braga, A. Lledos, G. Ujaque and F. Maseras, Acc. Chem. Res., 2013, 46, 2626 CrossRef CAS PubMed.
- J. Guerra and M. A. Herrero, Nanoscale, 2010, 2, 1390 RSC.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC.
- Z. Chen, Z.-M. Cui, F. Niu, L. Jiang and W.-G. Song, Chem. Commun., 2010, 46, 6524 RSC.
- J. Zhi, D. Song, Z. Li, X. Lei and A. Hu, Chem. Commun., 2011, 47, 10707 RSC.
- J. Sun, Z. Dong, X. Sun, P. Li, F. Zhang, W. Hu, H. Yang, H. Wang and R. Li, J. Mol. Catal. A: Chem., 2013, 367, 46 CrossRef CAS PubMed.
- Z. Chen, Z.-M. Cui, P. Li, C.-Y. Cao, Y.-L. Hong, Z.-Y. Wu and W.-G. Song, J. Phys. Chem. C, 2012, 116, 14986 CAS.
- M. M. Coulter, J. A. Dinglasan, J. B. Goh, S. Nair, D. J. Anderson and V. M. Dong, Chem. Sci., 2010, 1, 772 RSC.
- J.-P. Tessonnier, L. Pesant, G. Ehret, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2005, 288, 203 CrossRef CAS PubMed.
- M. Endo, Y. A. Kim, M. Ezaka, K. Osada, T. Yanagisawa, T. Hayashi, M. Terrones and M. S. Dresselhaus, Nano Lett., 2003, 3, 723 CrossRef CAS.
- Y.-J. Gu and W.-T. Wong, J. Nanosci. Nanotechnol., 2009, 9, 2066 CrossRef CAS PubMed.
- F. P. Zamborini, S. M. Gross and R. W. Murray, Langmuir, 2001, 17, 481 CrossRef CAS.
- B. Cornelio, G. A. Rance, M. Laronze-Cochard, A. Fontana, J. Sapi and A. N. Khlobystov, J. Mater. Chem. A, 2013, 1, 8737 CAS.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- M. Perez-Lorenzo, J. Phys. Chem. Lett., 2012, 3, 167 CrossRef CAS.
- R. Narayanan, C. Tabor and M. A. El-Sayed, Top. Catal., 2008, 48, 60 CrossRef CAS.
- A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Chem.–Eur. J., 2007, 13, 6908 CrossRef CAS PubMed.
- P.-P. Fang, A. Jutand, Z.-Q. Tian and C. Amatore, Angew. Chem., Int. Ed., 2011, 50, 12184 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, Top. Catal., 2008, 47, 5181 CrossRef.
- P. J. Ellis, I. J. S. Fairlamb, F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820 CrossRef CAS PubMed.
- A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473 RSC.
- J. G. de Vries, Dalton Trans., 2006, 421 RSC.
- D. Peral, F. Gomez-Villarraga, X. Sala, J. Pons, J. C. Bayon, J. Ros, M. Guerrero, L. Vendier, P. Lecante, J. Garcia-Anton and K. Philippot, Catal. Sci. Technol., 2013, 3, 475 CAS.
- L. Ghosez, C. Franc, F. Denonne, C. Cuisinier and R. Touillaux, Can. J. Chem., 2001, 79, 1827 CrossRef CAS.
- O. Navarro, H. Kaur, P. Mahjoor and S. P. Nolan, J. Org. Chem., 2004, 69, 3173 CrossRef CAS PubMed.
- O. Navarro, N. Marion, Y. Oonishi, R. A. Kelly III and S. P. Nolan, J. Org. Chem., 2006, 71, 685 CrossRef CAS PubMed.
- Z. Ahmadi and J. S. McIndoe, Chem. Commun., 2013, 49, 11488 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta06953f |
| This journal is © The Royal Society of Chemistry 2015 |